Cornelia de Lange syndrome is a syndrome of multiple congenital anomalies, characterized by a distinctive facial appearance, prenatal and postnatal growth deficiency, feeding difficulties, psychomotor delay, behavioral problems, and associated malformations that mainly involve the upper extremities. Classic cases of Cornelia de Lange syndrome are usually easy to diagnose, however mild cases may be challenging(1,2).
NEONATOLOGIE
Sindrom Cornelia de Lange asociat cu anomalie congenitală de cord. Prezentare de caz
Congenital heart disease in Cornelia de Lange syndrome. Case report
Ioana Roşca,
Anca Ristea,
Marcela Şerban,
Raluca Tocariu,
Viorica Tudor,
Mariana Nanea,
Mihai Mitran
First published: 15 octombrie 2015
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Gine.3.3.2015.4815
Abstract
Rezumat
Sindromul Cornelia de Lange este un sindrom plurimalformativ, caracterizat prin aspect tipic al feţei, retard de creştere intrauterină, dar şi postnatal, dificultăţi de alimentare, întârziere neuropsihică, tulburări de comportament şi malformaţii, care implică în special membrele superioare. Diagnosticul clinic este uşor de stabilit în formele tipice, dar poate fi dificil în formele mai uşoare(1,2).
Este prezentat cazul unui nou-născut, cu retard de creştere intrauterină, 37-38 de săptămâni de gestaţie, greutate la naştere 1.930 g, Scor Apgar 8 la 1 minut, care prezintă la naştere stare generală mediocră, tegumente cianotice, vernix abundent, bont ombilical subţire, normal inserat, microcefalie (PC=26 cm), cu suturi dehiscente, fontanela anterioară 3/2,5 cm, fontanela posterioară 2/2 cm, normotensive, respiraţii spontane, murmur vezicular prezent bilateral, AV=120 b/min, zgomote cardiace ritmice, suflu sistolic grad II-III/VI parasternal stâng, abdomen suplu, anus permeabil, absenţa degetului IV de la mâna dreaptă, sindactilie degete I-II picior drept, testicule ectopice, bilateral, tonus şi reactivitate bune, facies dismorfic, ţipăt de tonalitate joasă. Suspiciunea de anomalie genetică s-a ridicat încă din viaţa intrauterină, prin ultrasonografie morfologică fetală 4D, efectuată în dinamică.
1. Morfologie fetală 4D la VG = 22 săptămâni
Aspecte patologice:`
-
Biometric= 20,6 săptămâni.
-
Faţă cu profil anormal: os nazal prezent, hipoplazic, 4,8 mm, frunte cu oasele orbitei proeminente şi îngroşarea pielii la nivelul frunţii - 5 mm (figura 1); hipertelorism; buza superioară mult proeminentă, ce acoperă parţial buza inferioară (figura 2); frunte îngustă.
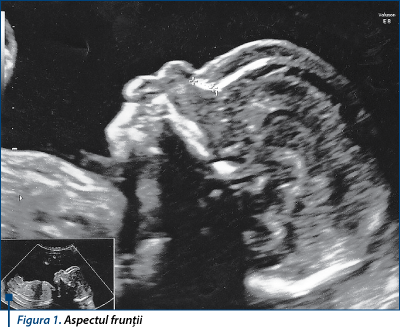
Figura 1
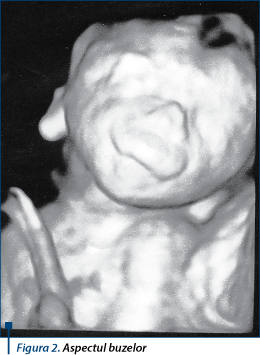
Figura 2 -
Regiune bucală normală.
-
Mâna dreaptă prezintă 4 degete, din care unul rudimentar - policele, arătătorul, degetul mijlociu rudimentar, inelar absent, degetul mic (figurile 3 şi 4).
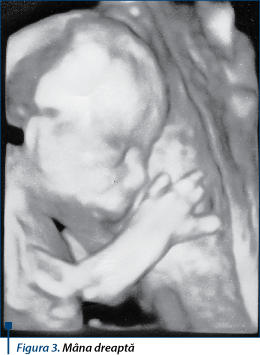
Figura 3
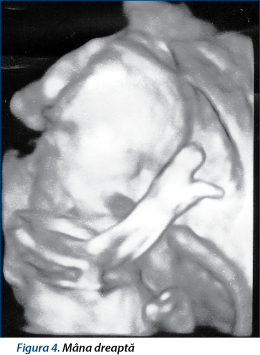
Figura 4 -
Mâna stângă cu 5 degete, fără mişcări de deschidere sau închidere şi este fixată în semiflexie.
-
Sex masculin.
-
Placenta anterioară, foarte subţire, grosime maximă = 1,5-2 cm, grad I, la distanţă de OCI.
-
LA în cantitate normală.
Concluzie: ecografic sunt depistate mai multe defecte structurale şi un grad de retard de creştere care ridică suspiciunea de sindrom genetic, mai ales trisomie 18 (sindrom Edwards).
Mama nu a efectuat teste de screening în primul trimestru de sarcină.
Părinţii sunt devotaţi sarcinii şi, în deplină cunoştinţă de cauză, doresc continuarea sarcinii, indiferent de anomaliile văzute ecografic.
2. Morfologie fetală 4D la VG = 29,3 săptămâni
-
Biometric = 27,1 săptămâni.
-
Se menţin defectele structurale descrise la morfologia de la 22 de săptămâni:
Aspect anormal al profilului feţei: bosa frontală, arcade frontale şi filtrum proeminente, micrognaţie uşoară, retrognaţie moderată-severă (figura 5).
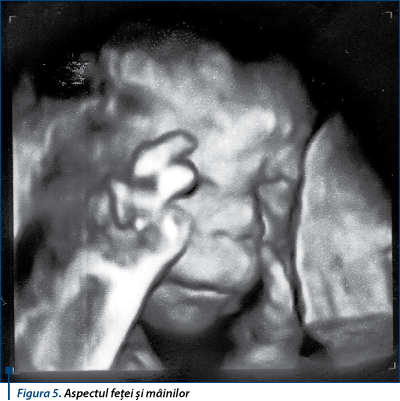
Aspect anormal al degetelor de la mâna dreaptă (figura 5), mâna stângă apare formată normal, fără mişcări la nivelul falangelor.
LA în cantitate crescută - AFI = 16,71.
Cord fetal asimetric (stâng<drept), cu Ao la nivelul arcului de 3,4 mm.
Concluzie: Suspiciune de sindrom Cornelia de Lange, dar nici cromozomopatiile de tip trisomie 13/18 nu pot fi excluse.
3. Morfologie fetală 4D la VG = 34,2 săptămâni
-
Biometric = 30,1 săptămâni.
-
Sunt vizibile toate defectele structurale descrise anterior.
-
Polihidramnios - AFI = 21,27.
Concluzie: restricţie de creştere de cauză fetală, probabil sindrom Cornelia de Lange.
După naştere se instituie tratament cu perfuzie de reechilibrare hidroelectrolitică şi acido-bazică, antibioterapie cu spectru larg (nou-născutul prezintă sindrom inflamator biologic important), oxigenoterapie sub cort cefalic, monitorizare cardiorespiratorie, monitorizarea diurezei şi a tensiunii arteriale.
În evoluţie se menţine oxigeno-dependent şi echilibrat hemodinamic. Este alimentat prin gavaj din ziua a treia de viaţă cu cantităţi progresiv crescute de formulă adaptată vârstei, cu toleranţă digestivă bună, însă prezintă un reflux gastroesofagian important, necesitând administrare de formulă antireflux. Nou-născutul nu prezintă reflex de supt şi nu se reuşeşte alimentarea acestuia la biberon.
Având în vedere aspectul nou-născutului, se menţine suspiciunea de sindrom Cornelia de Lange.
Ecografia transfontanelară (ziua întâi de viaţă):
-
Aparent fără malformaţii, structuri cerebrale simetrice faţă de linia mediană, ventriculi laterali normal conformaţi, fără hemoragie intra-periventriculară, corp calos prezent, plexuri coroide normal conformate, fără imagini hipo-/hiperecogene.
Consult neurologie pediatrică (ziua a patra de viaţă):
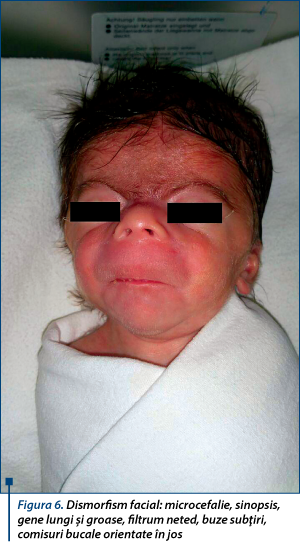
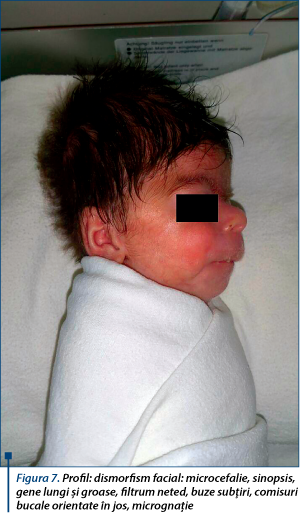
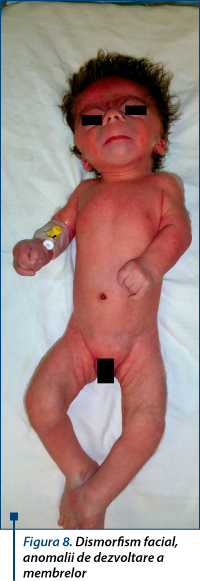
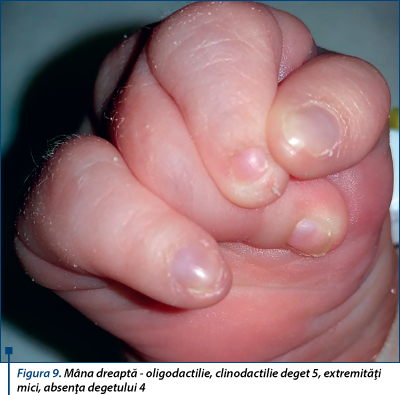
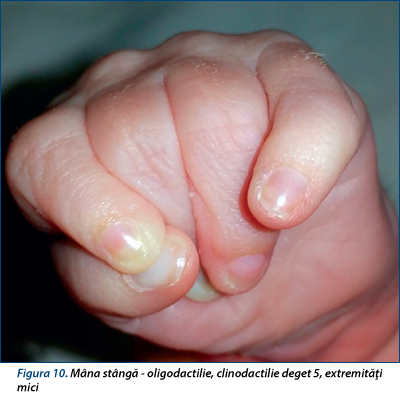
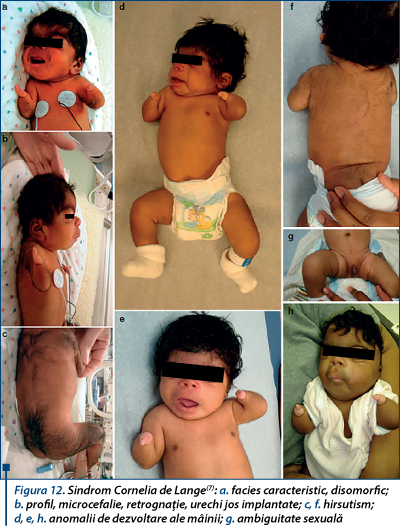
Nou-născut în vârstă de 4 zile cu restricţie de creştere intrauterină, fenotip sugestiv pentru sindrom Cornelia de Lange (figurile 6, 7, 8), prezintă la evaluare: motilitate spontană redusă, pumni închişi (figurile 9, 10), hipotonie axială, cap balant, nu îşi susţine greutatea, mers automat absent, Galant absent, nu se obţine reflex cohleo-palpebral, alimentat prin gavaj.
Concluzie: tulburare globală de dezvoltare, de cauze prenatale, genetice. Pentru episoadele paroxistice, cu clonii ale membrelor, s-a efectuat electroencefalograma, care are aspect normal pentru vârstă, fără elemente epileptice. Se recomandă temporizarea introducerii medicaţiei antiepileptice, însă, dacă persistă crizele, se recomandă încărcarea cu fenobarbital 10 mg şi o reevalure neurologică.
Consultul ORL (ziua a 16-a de viaţă):
Laringe cu aspect şi motilitate normale. Fără obstrucţie respiratorie mecanică la nivelul căilor aeriene.
Secreţii mucose relativ abundente faringiene şi la nivelul arborelui traheo-bronşic.
Concluzii: fără malformaţii traheo-bronşice.
Ecografia cardiacă evidenţiază defect septal interventricular perimembranos mic/moderat, stenoză pulmonară largă, persistenţă de foramen ovale.
Consult genetic:
Dismorfism facial: microcefalie, sinopsis, gene lungi şi groase, filtrum neted, buze subţiri, comisuri bucale orientate în jos, micrognaţie (figurile 6, 7, 8). Anomalii de dezvoltare a membrelor (oligodactilie, clinodactilie deget 5 membre superioare, sindactilie cutanată deget 1-2 picior drept, extremităţi mici, limitarea mişcărilor în articulaţia coatelor; figurile 9 şi 10), suflu sistolic, retard de creştere pre-/postnatal.
Tabloul clinic susţine diagnosticul de Sindrom Cornelia de Lange.
Evoluţia a fost lent favorabilă, probele inflamatorii negativate. A necesitat transfuzie cu sânge ales, la Institutul de Hematologie, 10 ml/kg corp, din cauza unei anemii plurietiologice, echilibrat cardiorespirator, SpO2=98% în aerul atmosferic, AV=140 b/min, suflu sistolic staţionar, abdomen suplu, tranzit intestinal prezent, toleranţă digestivă bună, fără reflex de supt, fiind alimentat prin gavaj discontinuu, scădere ponderală fiziologică, curbă ponderală ascendentă din a 10-a zi de viaţă, tonus şi reactivitate diminuate.
Greutatea actuală: 2.050 g (30 zile de viaţă).
Incidenţa sindromului Cornelia de Lange este de 1 caz la 10.000-50.000 de nou-născuţi. Nu există diferenţe de incidenţă în funcţie de sex(1).
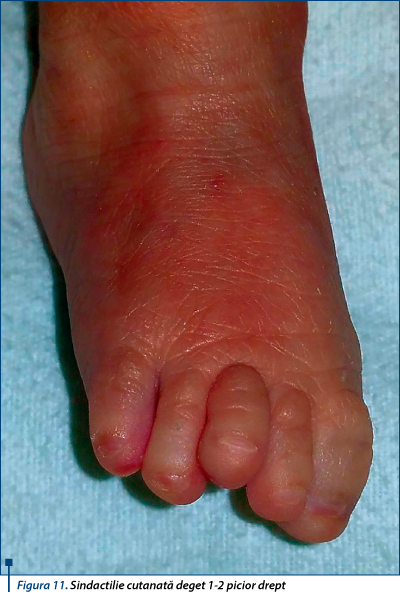
Diagnosticul prenatal poate fi stabilit prin identificarea ecografică a unor anomalii cunoscute ca fiind asociate cu acest sindrom: retard de creştere intrauterină, defecte ale membrelor, hernie diafragmatică, antebraţe hipoplazice, mâini cu dezvoltare anormală şi defecte faciale tipice. Diagnosticul prenatal prin tehnici de genetică moleculară este posibil dacă se cunoaşte mutaţia în cadrul familiei; totuşi, mai mult de 99% din cazuri sunt sporadice(3).
A fost descrisă rar transmiterea autozomal-dominantă sau x linkată în cadrul unei familii în care părinţii aveau forme uşoare ale acestui sindrom. Singurele gene asociate cu sindromul Cornelia de Lange sunt genele NIPBL de pe cromozomul 5 şi SMC1L1 pe cromozomul X(4).
Clinic, sindromul Cornelia de Lange se caracterizează prin retard de creştere, trăsături faciale distinctive, malformaţii ale mâinilor, picioarelor, braţelor, retard mental (figura 12).
Severitatea simptomatologiei variază de la caz la caz. Persoanele afectate prezintă retard de creştere atât intrauterin, cât şi postnatal.
Cei mai mulţi nou-născuţi prezintă greutate mică pentru vârsta gestaţională şi curbă ponderală nesatisfăcătoare în perioada de sugar. Copiii pot prezenta tulburări de alimentaţie prin afectarea deglutiţiei în primele luni sau ani de viaţă(2,5,6).
Alte manifestări prezente la copiii afectaţi sunt hipertonie şi plâns/ ţipăt specific de tonalitate joasă, microcefalie, gât scurt, linia de implantare a părului coborâtă, nas mic cu narine anteversate, sprâncene bine conturate, unite pe linia mediană, gene lungi curbate, buze subţiri cu comisurile bucale îndreptate inferior, filtrum nazal lung, micrognaţie, dinţii sunt de obicei mici, hipoplazici, cu spaţii mari între ei şi erupţie întârziată, urechi jos implantate. Se poate asocia boltă ogivală sau palatoschizis(2,5,6).
Malformaţiile membrelor includ clinodactilie, oligodactilie, poziţionare anormală a policelui, limitarea mişcărilor în articulaţia cotului, hipoplazia oaselor mâinii sau piciorului, membre scurte; în unele cazuri pot lipsi degete, mâna sau chiar antebraţul.
Afectarea membrelor superioare poate fi unilaterală sau bilaterală; dacă afectarea este bilaterală, aceasta poate fi asimetrică. Vârsta osoasă la aceşti copii este întârziată(1,2,7).
Retardul psihomotor poate fi de diferite grade, de la uşor la sever, şi poate fi însoţit de tulburări de comportament, precum episoade de agresivitate, cu loviri şi ţipete. Deşi acestor copii cel mai frecvent le lipseşte expresivitatea facială, par să răspundă pozitiv la anumiţi stimuli(2,7).
În multe cazuri, copiii cu sindrom Cornelia de Lange prezintă afectarea auzului şi întârziere în achiziţia limbajului. Sunt frecvente episoadele de otită medie, care uneori se cronicizează(1,2,6).
Aspectul tegumentelor copiilor afectaţi poate fi marmorat, cu discretă tentă cianotică periorificial. În plus, pot prezenta creşterea părului în exces pe anumite zone ale corpului, incluzând urechile, zona lombară şi membrele(1,2,6).
La nivel genitourinar, copiii afectaţi pot prezenta hipoplazia organelor genitale, criptorhidie, hipospadias(1,2).
Alte anomalii întâlnite în cadrul sindromului Cornelia de Lange sunt malformaţiile cardiace, susceptibilitate crescută pentru infecţii respiratorii, afectare oculară (miopie, nistagmus, ptoză palpebrală) şi uneori crize convulsive(1,6).
Complicaţiile gastrointestinale reprezintă cea mai frecventă cauză de deces la aceşti copii; acestea incluzând hernia diafragmatică şi pneumonia de aspiraţie(2).
Tratamentul constă în stimulare neuropsihică, monitorizare din partea unei echipe multidisciplinare: pediatru, cardiolog, genetician, neurolog, gastroenterolog, oftalmolog, tratament simptomatic al afecţiunilor asociate (convulsii, reflux gastroesofagian) şi corecţia chirurgicală a malformaţiilor asociate, acolo unde acestea există (palatoschizis, stenoză hipertrofică de pilor, criptorhidie)(1,2,7,8).
Prognosticul cazului
Este rezervat din punctul de vedere al retardului somatic, mental şi al asocierii cu malformaţie congenitală de cord, evoluţia pe termen lung depinzând de îngrijirea medicală, complianţa familiei şi răspunsul copilului la tratamentul multidisciplinar.
Coroborând datele actuale ale sugarului cu datele din literatura de specialitate, ne putem aştepta la următoarele posibilităţi de evoluţie:
-
internare de lungă durată sau internări repetate, întrucât sugarul prezintă un reflux gastroesofagian sever, fiind în tratament cu nexium şi motilium per os şi alimentat cu formulă antireflux;
-
câştig ponderal deficitar consecutiv refluxului gastroesofagian sever şi vărsăturilor asociate;
-
infecţii respiratorii recurente (din cauza refluxului gastroesofagian, precum şi a spitalizărilor prelungite/repetate);
-
infecţii sistemice şi urinare, fiind vorba de un sugar cu motilitate redusă şi internări repetate;
-
defectele cardiace se pot închide în timp sau pot necesita terapie chirurgicală. Aceste defecte pot accentua malnutriţia;
-
recuperarea neurologică (oricum dificilă) poate fi întârziată de patologia asociată (reflux gastroesofagian important, deficit ponderal, malformaţie cardiacă);
-
exitus în primul an de viaţă din cauza complicaţiilor: pneumonie de aspiraţie, internări de lungă durată cu infecţii repetate (familia nepregătită pentru îngrijirea sugarului la domiciliu).
În evoluţie, este necesară consultarea următorilor specialişti: geneticieni, cardiologi, oftalmologi, audiologi, neurologi. Se recomandă, de asemenea, logopedie, ergoterapie şi consiliere psihopedagogică(7,8).
Particularitatea cazului
Este un caz tipic de sindrom Cornelia de Lange, asociat cu malformaţie congenitală de cord (defect septal interventricular perimembranos mic/moderat, stenoză pulmonară largă, persistenţă de foramen ovale), ridicându-se suspiciunea, precoce, de anomalie genetică şi de malformaţie cardiacă, încă din viaţa intrauterină, prin ultrasonografie morfologică fetală 4D, efectuată seriat, începând cu a 22-a săptămână de gestaţie.
Bibliografie
Jones KL. Smith’s Recognizable Patterns of Human Malformation. 5th ed. Philadelphia, PA; W.B. Saunders Comp;1997:88-9.
Preus M, Rex AP. Definition and diagnosis of the Brachmann-de Lange syndrome. Am J Med Genet, 1983;16:301-12.
Ranzini AC, Day-Salvatore D, Farren-Chavez D, et al. Prenatal diagnosis of de Lange syndrome. J Ultrasound Med;1997:16.
Gil-Rodriguez MC, Ciero M, Lopez-Vinas E, Ribate MP, Arnedo M, Deardorff MA, Puisac B, Legarreta J, Karam JC, Rubio E, Bueno I, et al. Mutations and variants in the cohesion factor genes NIPBL, SMC1A, and SMC3 in a cohort of 30 unrelated patients with Cornelia de Lange syndrome. Am J Med Genet,2010;152A: 924-9.
Chatfield KC, Schrier SA, Li J, Clark D, Kaur M, Kline AD, Deardorff MA, Jackson LS, Goldmuntz E, Krantz ID. Congenital heart disease in Cornelia de Lange syndrome: Phenotype and genotype analysis, Am J Med Genet, part A,158A;2010: 2499–2505.
Gorlin RJ, et al. Syndromes of the Head and Neck, 3rd ed, NY, Oxf Univ Press; 1990:300-4.
www.rarediseases.org/rare-diseases/cornelia-de-lange-syndrome.
Russell KL, Ming JE, Patel K, et al. Dominant paternal transmission of Cornelia de Lange syndrome: a new case and review of 25 previously reported familial recurrences. Am J Med Genet, 2001;104(4):267-76.
Preus M, Rex AP. Definition and diagnosis of the Brachmann-de Lange syndrome. Am J Med Genet, 1983;16:301-12.
Ranzini AC, Day-Salvatore D, Farren-Chavez D, et al. Prenatal diagnosis of de Lange syndrome. J Ultrasound Med;1997:16.
Gil-Rodriguez MC, Ciero M, Lopez-Vinas E, Ribate MP, Arnedo M, Deardorff MA, Puisac B, Legarreta J, Karam JC, Rubio E, Bueno I, et al. Mutations and variants in the cohesion factor genes NIPBL, SMC1A, and SMC3 in a cohort of 30 unrelated patients with Cornelia de Lange syndrome. Am J Med Genet,2010;152A: 924-9.
Chatfield KC, Schrier SA, Li J, Clark D, Kaur M, Kline AD, Deardorff MA, Jackson LS, Goldmuntz E, Krantz ID. Congenital heart disease in Cornelia de Lange syndrome: Phenotype and genotype analysis, Am J Med Genet, part A,158A;2010: 2499–2505.
Gorlin RJ, et al. Syndromes of the Head and Neck, 3rd ed, NY, Oxf Univ Press; 1990:300-4.
www.rarediseases.org/rare-diseases/cornelia-de-lange-syndrome.
Russell KL, Ming JE, Patel K, et al. Dominant paternal transmission of Cornelia de Lange syndrome: a new case and review of 25 previously reported familial recurrences. Am J Med Genet, 2001;104(4):267-76.