The careful monitoring of nutritional status is crucial for patients with cystic fibrosis (CF). As part of routine CF care, the measurement of weight and height (and calculation of weight/length or of Body Mass Index – as appropriate) should be carried out and analyzed at every medical visit. The early recognition of nutritional risk factors is imperative, as well as the evaluation of the patient’s condition by a multidisciplinary team in order to identify the caloric intake, the risk of malabsorption and other risk factors for growth deficit. Along with the usual tools of nutritional intervention to improve the nutritional status of patients with CF, oral supplementation, interventions to correct eating disorders, appetite suppressant supplements/medication and enteral nutrition are also included.
Nutriţia pacientului cu fibroză chistică în contextul îngrijirii multidisciplinare
Nutrition of the patient with cystic fibrosis in the context of multidisciplinary care
First published: 23 decembrie 2020
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Med.138.6.2020.3999
Abstract
Rezumat
Monitorizarea atentă a stării nutriţionale este esenţială pentru pacienţii cu fibroză chistică (FC). Ca parte a îngrijirii de rutină a FC, măsurarea greutăţii şi a înălţimii (şi calculul greutăţii/lungimii sau al indicelui de masă corporală – după caz) trebuie efectuată şi analizată cu ocazia fiecărei vizite medicale a pacientului. Recunoaşterea timpurie a factorilor de risc nutriţional este imperativă, precum şi evaluarea stării pacientului de către o echipă multidisciplinară, în vederea identificării aportului caloric, a riscului de malabsorbţie şi a altor factori de risc pentru deficitul de creştere. Alături de instrumentele obişnuite de intervenţie nutriţională pentru îmbunătăţirea statusului nutriţional al pacienţilor cu FC, se regăsesc şi suplimentarea orală, intervenţiile de corectare a tulburărilor de comportament alimentar, suplimentele/medicaţia stimulatoare de apetit şi alimentaţia enterală.
Monitorizarea atentă a nutriţiei şi creşterii este esenţială în îngrijirea adulţilor şi a pacienţilor pediatrici cu fibroză chistică (FC)(1). Fundaţia pentru Fibroză Chistică recomandă ca monitorizarea stării nutriţionale să fie parte integrantă în îngrijirea de rutină a FC, fiind de dorit ca atât adulţii, cât şi copiii cu FC să aibă un status nutriţional comparabil cu al celor sănătoşi, ceea ce va contribui la o evoluţie clinică mai bună(2,3). Cu toate acestea, starea nutriţională optimă poate fi greu de realizat şi de menţinut la aceşti pacienţi.
Pacienţii cu FC şi familiile lor ar trebui să fie educaţi cu privire la importanţa îngrijirii nutriţionale pe tot timpul vieţii(4). Se recomandă revizuirea istoricului dietei la fiecare 3-6 luni(5). În situaţiile în care un pacient nu este capabil să atingă obiectivele nutriţionale stabilite, intervenţiile nutriţionale trebuie implementate devreme, fiind necesar ca toate opţiunile să fie aduse la cunoştinţa pacientului şi/sau a familiei sale. Este important ca aceştia să fie informaţi asupra variantelor existente pentru îmbunătăţirea stării nutriţionale, astfel încât să fie pe deplin implicaţi în luarea deciziilor privind managementul bolii. Trebuie ţinut cont de faptul că, de cele mai multe ori, atât pacienţii, cât şi familiile lor au nevoie de timp pentru a accepta şi a ajunge să se simtă confortabil cu utilizarea unor intervenţii invazive cum este alimentaţia enterală(6).
Evaluarea iniţială a copilului cu risc nutriţional trebuie să includă determinarea aportului caloric (istoricul dietei), să ţină cont de prezenţa malabsorbţiei (prezenţa steatoreei, istoricul pancreatic, administrarea de enzime) şi dacă există o exacerbare a afectării pulmonare(1,7). Iniţial se abordează cauzele identificabile(1), cum ar fi aportul caloric insuficient, care poate fi diagnosticat de către dietetician pe baza unui jurnal alimentar. Fiecare pacient este sfătuit să îşi maximizeze aportul de energie(1,7), iar recomandările dietetice sunt adaptate individual(5). Consilierea nutriţională ar trebui făcută de către un dietetician experimentat, cu încurajarea unei diete bogate în grăsimi(8) mononesaturate şi polinesaturate în detrimentul celor saturate(9), respectiv o dietă alcătuită în principal din alimentele bogate în acid linoleic şi alţi acizi graşi polinesaturaţi cu lanţ lung(5).
Pentru pacienţii cu risc nutriţional se recomandă abordarea multidisciplinară, cu ajutorul unei echipe formate din pneumolog, dietetician, gastroenterolog, endocrinolog, psiholog, asistent medical, asistent social, fiziochinetoterapeut, specializaţi în CF, şi medic de familie(7,4). Pentru aceşti pacienţi, investigaţiile includ analiza şi a altor comorbidităţi care pot contribui la starea nutriţională suboptimală. În acest sens, este nevoie de evaluarea de către gastroenterolog a cauzelor gastrointestinale legate de FC, care contribuie la reducerea apetitului şi/sau la pierderi intestinale crescute, inclusiv malabsorbţie, populare bacteriană la nivelul intestinului subţire, boala de reflux gastroesofagian, constipaţia şi afectarea hepatică legată de FC, precum şi evaluarea riscului pentru alte boli gastrointestinale, cum sunt gastroenterita eozinofilică, boala celiacă, tulburările de motilitate intestinală şi boala inflamatorie intestinală(10). Scăderea toleranţei la glucoză şi diabetul zaharat, frecvente în FC, pot, de asemenea, contribui la starea nutriţională deficitară, ceea ce necesită şi implicarea unui diabetolog în echipa de îngrijire a pacientului cu FC(1,7).
Rolul psihologilor şi al psihiatrilor este esenţial în echipa multidisciplinară, deoarece pot evalua cauzele psihosociale a stării nutriţionale suboptimale, inclusiv stările depresive, anxioase, tulburările de imagine corporală şi anorexia nervoasă(4,5,7). Asistenţilor sociali le revine rolul de a evalua problemele de ordin financiar şi alţi factori de stres social care pot contribui la un status nutriţional slab(4,7). Cauzele de deficit nutriţional mai frecvent întâlnite la pacienţii cu CF sunt sintetizate în tabelul 1.
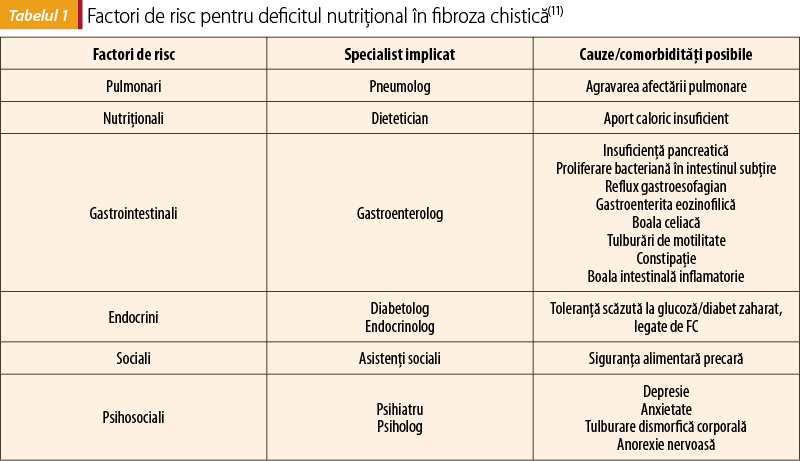
Un comportament alimentar dezordonat poate afecta starea nutriţională la copiii mici şi la cei de vârstă preşcolară cu FC, la care problemele de comportament alimentar sunt frecvent întâlnite şi pot interfera cu atingerea statusului nutriţional optim(12-14). Terapia orientată spre comportamentul alimentar s-a dovedit a fi mai eficientă decât îngrijirea nutriţională standard în ceea ce priveşte îmbunătăţirea aportului caloric general şi a scorului Z al înălţimii(15). La adolescenţii şi adulţii cu FC, percepţia proprie eronată despre imaginea corporală poate afecta negativ aportul nutriţional(16,17). Este important ca aceşti factori să fie identificaţi şi trataţi, dacă sunt prezenţi.
Pentru adulţii şi copiii cu status nutriţional suboptimal şi aport caloric insuficient, pot fi recomandate stimulatoare de apetit în vederea îmbunătăţirii aportului caloric. Cu toate că această suplimentare este recomandată în ghidurile Fundaţiei pentru Fibroză Chistică, Societatea Europeană pentru Nutriţie Clinică şi Metabolism (ESPEN) nu a oferit recomandări bazate pe dovezi, din cauza datelor insuficiente din literatura de specialitate(5,7). Cel mai frecvent recomandate stimulatoare de apetit sunt ciproheptadina, acetatul de megestrol şi dronabinolul. Ciproheptadina este un antihistaminic cu efect secundar de stimulare a apetitului. Pentru pacienţii cu insuficienţă pancreatică şi stare nutriţională optimă, administrarea de enzime pancreatice se recomandă ca parte a tratamentului de rutină(18).
Terapiile nutriţionale specifice prin diete cu aport mai crescut de sodiu, zinc şi acizi graşi esenţiali au fost, de asemenea, cercetate pentru a identifica măsura în care acestea pot contribui la corectarea unui status nutriţional suboptimal(11).
Suplimentarea cu sodiu este esenţială la pacienţii cu FC, deoarece pierderea excesivă de sare prin piele apare frecvent, ceea ce poate duce la deshidratare hiponatremică, mai ales la sugari, şi la încetinirea câştigului ponderal(19). În consecinţă, suplimentarea cu sodiu (sub formă de clorură de sodiu) la sugarii sub 6 luni presupune 0,6 g de clorură de sodiu zilnic, iar la sugarii de 6 luni şi peste (şi copiii cu o dietă săracă în sare) ar trebui să se administreze 1,2 g de clorură de sodiu zilnic(7,20). În plus, copiii mai mari şi adulţii din zonele calde sau cei care depun efort în timpul exerciţiilor fizice necesită, de asemenea, suplimentare. Se recomandă ca băuturile pentru sportivi să conţină 0,6 g de clorură de sodiu la 350 ml de băutură pentru a evita hiponatremia la cei cu FC(21).
Ghidurile ESPEN sugerează adăugarea a 1-2 mmol/kg corp/zi de sodiu la sugarii alăptaţi, până la 4 mmol/kg corp/zi la sugarii cu pierderi crescute de sodiu şi aport de alimente sărate la copiii mai mari şi la adulţi(5).
Zincul este un micronutrient important pentru creştere şi imunitate, iar pacienţii cu FC prezintă risc de deficit de zinc, deoarece malabsorbţia grăsimilor poate afecta negativ absorbţia zincului(22). Având în vedere că deficienţa de zinc poate duce la o întârziere a creşterii, Fundaţia pentru Fibroză Chistică recomandă suplimentarea cu zinc timp de 6 luni (1 mg zinc elementar/kg corp/zi în doze divizate) la sugari şi copii cu deficit de creştere(1,20). Recomandările ESPEN sugerează suplimentarea cu zinc pentru pacienţii cu FC care prezintă risc pentru deficit de zinc(5).
Aportul de acizi graşi esenţiali (AGE) este asigurat prin dietele bogate în uleiuri vegetale şi peşte de apă rece. Acidul alfa-linolenic este metabolizat în continuare în acid eicosapentanoic (EPA) şi acid docosahexanoic (DHA), în timp ce acidul linoleic este metabolizat în continuare în acid arahidonic şi acid dihomogama-linolenic. Deficienţa de AGE, mai frecvent întâlnită la bolnavii cu FC şi cu insuficienţă pancreatică, poate genera retard de creştere, dermatită, alopecie şi/sau trombocitopenie, de aceea se recomandă tratamentul deficienţei de AGE la cei care prezintă o creştere în greutate sub cea optimă(1,20).
Terapia antioxidantă, în special cu glutation, este în curs de cercetare, urmărindu-se efectul glutationului asupra nutriţiei şi creşterii la copiii şi adolescenţii cu FC(11).
Unele studii efectuate la pacienţii cu FC au demonstrat îmbunătăţirea parametrilor de creştere (înălţime, greutate şi masă musculară) în urma administrării hormonilor de creştere(23), iar alte studii au demonstrat îmbunătăţirea funcţiei pulmonare la administrarea hormonilor de creştere, dar mai sunt necesare în continuare studii pe o perioadă mai lungă, înainte de introducerea în tratamentul de rutină a hormonilor de creştere la pacienţii cu FC(23).
Utilizarea nutriţiei enterale, în special suplimentarea prin intermediul sondei nazogastrice, sondei gastrostomice sau gastrojejunale, este un instrument important în gestionarea insuficienţei nutriţionale la pacienţii cu FC, deoarece alimentaţia enterală îmbunătăţeşte statusul nutriţional şi de creştere în cazul copiilor(8,24-30). Nutriţia enterală ar trebui luată în considerare ca opţiune oricând pe tot parcursul vieţii unui pacient cu FC şi nu trebuie privită ca o „ultimă soluţie”(4).
Fundaţia pentru Fibroză Chistică recomandă utilizarea alimentaţiei enterale atunci când valorile parametrilor antropometrici nu pot fi îmbunătăţiţi, respectiv nu poate fi asigurat aportul caloric, în ciuda abordării multidisciplinare(4).
ESPEN recomandă iniţierea hrănirii enterale în cazul unui eşec persistent de creştere(5). Informarea şi educarea din timp a pacienţilor cu FC şi a aparţinătorilor privind beneficiile alimentaţiei enterale au rolul de a reduce disconfortul resimţit de aceştia faţă de alimentaţia enterală, atunci când aceasta devine necesară(4,24,31).
La sugarii cu FC, laptele matern (dacă este disponibil) şi formulele standard pentru sugari sunt cele utilizate în alimentaţie, cu excepţia cazului în care pacienţii prezintă intoleranţă. Densitatea calorică crescută a aportului de hrană este frecvent recomandată, pentru a obţine o stare nutriţională mai bună(4,20).
Există, de asemenea, dovezi cu privire la terapia de substituţie cu enzime pancreatice în timpul alimentaţiei enterale(4), cea mai comună metodă de administrare fiind înainte şi după administrarea formulelor enterale(5,32).
Nutriţia parenterală trebuie utilizată doar în cazul în care nutriţia enterală este contraindicată (rezecţie intestinală, sugar cu ileus de meconiu sau alte cauze ale insuficienţei intestinale)(5,33), fiind frecvent asociată unor riscuri semnificative(34).
Concluzii
Îngrijirea pacienţilor cu fibroză chistică necesită o echipă multidisciplinară specializată, esenţială pentru recunoaşterea celor cu risc de insuficienţă nutriţională şi pentru identificarea cauzelor potenţiale de malnutriţie sau deficit de creştere. Rolul dieteticianului în echipa de îngrijire este determinant mai ales pentru monitorizarea de rutină a creşterii şi a stării nutriţionale. Intervenţia nutriţională agresivă (inclusiv hrănirea prin intermediul tubului enteral) poate fi esenţială pentru a ajuta pacienţii să atingă obiectivele nutriţionale stabilite şi care se încadrează în recomandările ghidurilor de specialitate.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Borowitz D, Baker RD, Stallings V. Consensus report on nutrition for pediatric patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2002;35(3):246–59.
-
Yen EH, Quinton H, Borowitz D. Better nutritional status in early childhood is associated with improved clinical outcomes and survival in patients with cystic fibrosis. J Pediatr. 2013;162(3):530-5 e1.
-
Steinkamp G, Wiedemann B. Relationship between nutritional status and lung function in cystic fibrosis: cross sectional and longitudinal analyses from the German CF quality assurance (CFQA) project. Thorax. 2002;57(7):596-601.
-
Schwarzenberg SJ, et al. Enteral tube feeding for individuals with cystic fibrosis: evidence-informed guidelines. J Cyst Fibros. 2016;15(6):724–35.
-
Turck D, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin Nutr. 2016;35(3):557–77.
-
White H, et al. Enteral tube feeding in adults with cystic fibrosis; patient choice and impact on long term outcomes. J Cyst Fibros. 2013;12(6):616-22.
-
Lahiri T, et al. Clinical practice guidelines from the Cystic Fibrosis Foundation for preschoolers with cystic fibrosis. Pediatrics. 2016;137(4).
-
Stapleton D, Ash C, King S, Volders E, Graham C, Herd K, et al. Australasian clinical practice guidelines for nutrition in cystic fibrosis. D.A.O. Australia. 2006, Editor.
-
Brownlee J. Macronutrient requirements. In: Yen EH, editor. Nutrition in cystic fibrosis. Switzerland: Springer International Publishing. 2015;pp. 11-34.
-
Bahmanyar S, et al. Cystic fibrosis gene mutations and gastrointestinal diseases.
-
J Cyst Fibros. 2010;9(4):288-91.
-
Sullivan JS, Mascarenhas MR. Nutrition: Prevention and management of nutritional failure in Cystic Fibrosis. Journal of Cystic Fibrosis. 2017;16:S87-S93.
-
Stark LJ, et al. Parent and child mealtime behavior in families of children with cystic fibrosis. J Pediatr. 2000;136(2):195–200.
-
Powers SW, et al. Caloric intake and eating behavior in infants and toddlers with cystic fibrosis. Pediatrics. 2002;109(5):E75-.
-
Sanders MR, et al. Mealtime behavior and parent-child interaction: a comparison of children with cystic fibrosis, children with feeding problems, and nonclinic controls. J Pediatr Psychol. 1997;22(6):881–900.
-
Powers SW, et al. Behavioral and nutritional treatment for preschool-aged children with cystic fibrosis: a randomized clinical trial. JAMA Pediatr. 2015;169(5):e150636.
-
Truby H, Paxton AS. Body image and dieting behavior in cystic fibrosis. Pediatrics. 2001;107(6):E92.
-
Abbott J, et al. Nutritional status, perceived body image and eating behaviours in adults with cystic fibrosis. Clin Nutr. 2007;26(1):91-9.
-
Singh VK, Schwarzenberg SJ. Pancreatic insufficiency in Cystic Fibrosis. J Cyst Fibros. 2017;16(S2):S70-8.
-
Bower TR, Pringle KC, Soper RT. Sodium deficit causing decreased weight gain and metabolic acidosis in infants with ileostomy. J Pediatr Surg. 1988;23(6):567-72.
-
Cystic Fibrosis Foundation, et al. Cystic Fibrosis Foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. 2009;155(6 Suppl):S73-93.
-
Brownlee J. Macronutrient requirements. In: Yen EH, editor. Nutrition in cystic fibrosis. Switzerland: Springer International Publishing. 2015, pp. 11–34.
-
Krebs NF, et al. Low plasma zinc concentrations in young infants with cystic fibrosis. J Pediatr. 1998;133(6):761–4.
-
Thaker V, et al. Recombinant growth hormone therapy for cystic fibrosis in children and young adults. Cochrane Database Syst Rev. 2015;5:CD008901.
-
White H, et al. Enteral tube feeding in adults with cystic fibrosis; patient choice and impact on long term outcomes. J Cyst Fibros. 2013;12(6):616-22.
-
Vandeleur M, Massie J, Oliver M. Gastrostomy in children with cystic fibrosis and portal hypertension. J Pediatr Gastroenterol Nutr. 2013;57(2):245-7.
-
Bradley GM, et al. Nutritional outcomes following gastrostomy in children with cystic fibrosis. Pediatr Pulmonol. 2012;47(8):743-8.
-
Oliver MR, et al. Factors affecting clinical outcome in gastrostomy-fed children with cystic fibrosis. Pediatr Pulmonol. 2004;37(4):324-9.
-
Rosenfeld M, et al. Nutritional effects of long-term gastrostomy feedings in children with cystic fibrosis. J Am Diet Assoc. 1999;99(2):191-4.
-
Akobeng AK, Miller V, Thomas A. Percutaneous endoscopic gastrostomy feeding improves nutritional status and stabilizes pulmonary function in patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 1999;29(4):485-6.
-
Williams SG, et al. Percutaneous endoscopic gastrostomy feeding in patients with cystic fibrosis. Gut. 1999;44(1):87-90.
-
Gunnell S, et al. Attitudes toward percutaneous endoscopic gastrostomy placement in cystic fibrosis patients. J Pediatr Gastroenterol Nutr. 2005;40(3):334-8.
-
ATLAS Collaboration, et al. Measurement of kT splitting scales in W→ℓν events at (Formula: see text) with the ATLAS detector. Eur Phys J C Part Fields. 2013;73(5):2432.
-
Smyth AR, et al. European cystic fibrosis society standards of care: best practice guidelines. J Cyst Fibros. 2014;13(Suppl. 1):S23-42.
-
Allen ED, et al. Prolonged parenteral nutrition for cystic fibrosis patients. Nutr Clin Pract. 1995;10(2):73-9.
Articole din ediţiile anterioare
Prevalenţa şi validarea adicţiei alimentare în lumea contemporană
Termenul de adicţie este utilizat pentru a descrie o tulburare de consum de substanţe. În esenţă, aceasta reprezintă o afecţiune a creierului prima...
Medicina culinară – importanţă şi beneficii
Medicina culinară este un domeniu nou al medicinei bazate pe dovezi, ce îmbină hrănirea cu arta gătitului şi cu ştiinţa medicinei. Este destinată...