Eosinophilia is a frequent laboratory abnormality, associated with many hematological malignancies, solid tumors or with autoimmune diseases. It is most often found in Hodgkin lymphoma and in T-cell lymphoma. In T-cell lymphoma or adult’s T-cell leukemia, it is highly correlated with the peripheral leukemic cell count, disease extension and with Ann-Arbor stage. Hypereosinophilic syndrome (HES) is a heterogeneous group of clonal disorders characterized by significant eosinophilia without an underlying cause, and with clinical features due to eosinophilic infiltration of lungs, heart or serous membranes. HES can precede lymphoma’s onset, being associated with a poor prognosis. The paraclinical investigations are necessary to determine the organ damage and various cytogenetic and molecular assays are employed to identify rearrangements of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2 gene fusion. The upcoming case report describes a patient with a non-Hodgkin T-cell lymphoma associating HES, treated with multiple chemotherapeutic regimens. After each cycle, he obtained an unstable response. It was considered a highly aggressive disease with a poor prognosis and a severe evolution.
Hipereozinofilia în limfoamele maligne non-hodgkiniene T-periferice – mai mult decât o modificare hematologică
Hypereosinophilia in an aggressive peripheral T non-Hodgkin lymphoma – more than a hematological issue
First published: 20 decembrie 2021
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/OnHe.57.4.2021.5803
Abstract
Rezumat
Eozinofilia este o modificare hematologică frecventă, asociată cu neoplaziile hematologice, cu tumorile solide sau cu boli autoimune. Este cel mai frecvent întâlnită în limfoamele Hodgkin şi în limfoamele maligne non-hodgkiniene (LMNH) cu celulă T. În cadrul limfoamelor sau al leucemiei cu celulă T a adultului, se corelează cu nivelul celulelor leucemice din sângele periferic, cu extensia bolii şi cu stadiul Ann-Arbor. Sindromul hipereozinofilic (SHE) asociază afectare de organ (cord, plămân, seroase), precedând apariţia limfoamelor, fiind astfel un marker de prognostic negativ. SHE reprezintă un grup heterogen de boli clonale, caracterizate prin eozinofilie periferică marcantă şi manifestări clinice determinate de infiltratul eozinofilic la nivelul organelor şi ţesuturilor, fără o cauză de eozinofilie secundară. Paraclinic, sunt necesare investigaţii pentru a determina afectarea de organ. Examenele citogenetice şi moleculare pentru detectarea mutaţiilor PDGFRA, PDGFRB, FGFR1 şi PCM1-JAK2 sunt esenţiale pentru diagnostic. Prezentăm cazul unui pacient cu limfom malign non-hodgkinian cu celulă T cu SHE asociat, care a efectuat multiple linii de chimioterapie, după fiecare obţinând un răspuns hematologic care nu a fost însă menţinut, conturându-se într-o boală înalt agresivă, pacientul având un prognostic rezervat şi o evoluţie rapid nefavorabilă.
Introduction
Eosinophilia is a frequent laboratory abnormality, associated with hematologic malignancies such as Hodgkin lymphoma, non-Hodgkin lymphoma, myeloproliferative neoplasms, acute leukemia, as well as with non-hematologic neoplasms and autoimmune diseases(1). Both blood and bone marrow eosinophilia are a consequence of the secretion of interleukin-3, interleukin-5 and granulocyte-monocyte colony-stimulating factor, the main eosinophil growth and survival factors. These cytokines are produced not only by lymphoma cells, but also by normal lymphocytes stimulated by the neoplastic cells(2).
Eosinophilia occurrence depends on histological type of lymphoma, being encountered more often in T-cell lymphoma and Hodgkin lymphoma, and rarely in B-cell lymphoma(1). T-cell lymphoma may be accompanied by a variable degree of eosinophilia that correlates with peripheral leukemic cell counts, disease extension and with Ann-Arbor stage(1,2). In peripheral T-cell lymphoma, bone marrow eosinophilia is associated with a poor disease prognosis(1).
Hypereosinophilic syndrome (HES) is a heterogeneous group of rare disorders characterized by significant blood eosinophilia (>1500/mm3) without an underlying cause, and with clinical features due to eosinophilic infiltration of lungs, heart or serous membranes. HES can precede lymphoma’s onset, being associated with a poor prognosis(3).
The first step in the diagnostic algorithm is ruling out any secondary cause of eosinophilia, such as parasitic infections, allergies, drug hypersensitivity, autoimmune diseases (Churg-Strauss syndrome, Wegener granulomatosis, systemic lupus erythematosus), and the idiopathic disorder characterized by abnormal accumulation of eosinophils in lungs – chronic eosinophilic pneumonia. Secondly, paraclinical investigations are needed to identify the organ damage: hepatic enzymes, creatine kinase levels, troponin levels, renal function tests, electrocardiography and echocardiography, pulmonary function tests, and imagistic evaluation (computed tomography of the thorax, abdomen and pelvis)(3,4). Various cytogenetic and molecular assays are employed to identify rearrangements of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2 gene fusion(3,4).
Case presentation
We report the case of a 46-year-old male patient who was admitted to the Hematology Department of the Bucharest University Emergency Hospital, Romania, due to enlarged peripheral lymph nodes and fever for the past 14 days. He had also displayed generalized erythroderma for approximately five years, for which multiple biopsies had been done, without the histopathological confirmation of a hematological disease. He was previously treated with dexamethasone, methotrexate, antihistamines and photochemotherapy (PUVA). The response was initially favorable, but it was not maintained over time.
The clinical examination revealed generalized erythroderma, facial intradermal nodules, enlarged peripheral lymph nodes with a maximum diameter of 3 cm, and hepatosplenomegaly.
The laboratory tests indicated significant leukocytosis with important eosinophilia (WBC=100.600/mm3, EO=48%), increased LDH levels (LDH=1783 U/L) and mild hypofibrinogenemia (Fbg=185.60 mg/dl). The peripheral blood smear also was consistent with eosinophilia (EO=62%) in different maturation stages.
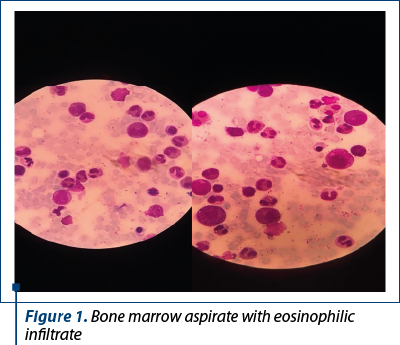
The bone marrow tap showed a hypercellular bone marrow with an important eosinophilic infiltrate (approximately 63%) and eosinophils in different maturation stages, some of them having immature basophilic granules.
The histopathological examination of lymph node biopsy revealed a diffuse malignant lymphoid proliferation with enlarged and medium size cells. Immunohistochemistry identified tumor cells positive for CD3 and CD30 (approximately 70%), with focal expression of CD25 and negative for CD 20 (B-cell marker) and ALK (anaplastic lymphoma kinase).
The diagnosis of non-Hodgkin lymphoma with large and medium T-cell was established.
Bone marrow biopsy highlighted a minimum bone marrow infiltrate (<10%) of lymphoma T-cells.
CT-scan of the thorax, abdomen and pelvis described generalized enlarged (supradiaphragmatic and subdiaphragmatic) lymph nodes (with maximum diameter 33 mm – celiac region), important hepatomegaly and mild splenomegaly.
The patient was tested for the FIP1L1-PDGFRA rearrangement, with a negative result.
Chemotherapy based on CHOEP protocol (cyclophosphamide, doxorubicin, etoposide, vincristine and prednisone) was initiated. After four cycles, the clinical features, leukocytosis with eosinophilia (WBC=71.000/mm3, EO=40.2%) were persistent, accompanied by severe hypofibrinogenemia (Fbg=78.27mg/dl). The imagistic work-up also described disease progression. Being considered a highly aggressive disease, the second-line therapy with brentuximab was initiated.
After three courses of brentuximab therapy, the eosinophil counts persisted at high levels, along with elevated lactate dehydrogenase activity (20-fold higher than the upper reference limit), progressive adenopathy on the CT-scan, bilateral pleural effusion, ascites in small quantity, and significant splenomegaly.
The molecular examination targeting the FIP1L1-PDGFRA rearrangement was repeated, this time providing a positive result.
A new diagnosis was established: stage IVB T-cell non-Hodgkin lymphoma, high risk IPI/PTCL-U score associating FIP1L1-PDGFRA positive hypereosinophilic syndrome – refractory disease after two treatment regimens (chemotherapy and immunotherapy), four cycles of CHOEP and three administrations of brentuximab.
DHAP (dexamethasone, cisplatin, cytarabine) salvage chemotherapy protocol was initiated. After the first day of treatment, the patient developed heart failure, most likely due to a volume overload, and cardiac dysfunction caused by the eosinophilic infiltration. This complication subsided under cardiologic treatment. The patient had also diarrhea, with no evidence of Clostridium difficile infection, pointing out to digestive tract involvement in HES.
Due to the highly resistant lymphoma and the modified molecular landscape suggestive for a clonal nature of HES, we initiated the GemDOx protocol (gemcitabine, dexamethasone, oxaliplatin) with imatinib, followed by a rapid clinical and biological improvement. However, two weeks later the patient developed significant asthenia, polypnea hypotension, edema, bilateral compressive enlarged inguinal lymph nodes, bilateral pleurisy and progressive tumoral hepatosplenomegaly. The laboratory findings revealed mild leukocytosis with eosinophilia (WBC=18,800/mm3, E=18%) and severe hypoglycemia (48 mg/dl). The patient was hospitalized in the hematology clinic in emergency condition. His evolution was quickly unfavorable, with persistent severe hypoglycemia which could not be corrected by 33% glucose infusions. The severe hypoglycemia is most probably explained by adrenal failure, caused either by the eosinophilic or lymphomatous infiltration, or by the corticosteroid treatment. Further, hemodynamic instability developed and it could not be restored by dopamine infusion. There were no clinical and biological signs for a septic shock. Therefore, the patient was transferred into the intensive care unit (ICU) with progressive disease and multiple organ dysfunction syndrome, with a rapid degrading status and the evolution of the case towards exitus.
Discussion
T-cell non-Hodgkin lymphoma represents a highly aggressive lymphoproliferative disease. It may particularly associate HES(1). Our patient was initially diagnosed with T-cell lymphoma, without associated HES, being negative for FIP1L1-PDGFRA rearrangement. Earlier, the patient received CHOEP regimen; however, disease progression was registered. Next, a second-line therapy with brentuximab was employed. After three courses of brentuximab, no clinical or biological improvement was obtained. Testing for FIP1L1-PDGFRA rearrangement was repeated, a positive result being provided this time. Upon these results, a salvage therapy was initiated – DHAP (dexamethasone, cisplatin, cytarabine), whilst the patient presented adverse events such as heart failure with systolic dysfunction and diarrhea. The protocol was switched to a GemDOx regimen with associated imatinib, a therapy that offered a short-term benefit for the patient. Two weeks after the GemDOx treatment, the patient developed adrenal insufficiency, hemodynamic instability and became comatose, being admitted to the ICU, where his condition degraded and led to exitus.
In our patient’s case, there is a high probability that the associated HES was the one key factor which led to an aggressive disease evolution. He was been refractory to four chemotherapy cycles, every time obtaining a partial response (clinical, biological, even reducing the eosinophils percent) for a short period, not maintaining the response over time. The patient also developed complications: heart failure with moderate systolic dysfunction, most likely considered to be the result of a combination of volume overload and cardiac eosinophilic infiltration, as well as diarrhea due to digestive tract damage (HES associated).
Conclusions
Eosinophilia occurrence is related to the histological type of lymphoma, being more frequently encountered in T-cell lymphomas, Hodgkin lymphoma, and rarely in B-cell lymphoma.
In literature it is well documented that HES can precede the diagnosis of non-Hodgkin lymphoma. Many experts consider the lymphoid type of HES a particular type of reactive HES, characterized by an increased eosinophilic production, caused by a group of abnormal circulant T-cells. T-cell lymphoma/leukemia associates eosinophilic levels which are correlated with the peripheral leukemic cell counts, disease extension and with Ann-Arbor stage(1,2).
When present in peripheral T-cell lymphoma, bone marrow eosinophilia is associated with a poor disease outcome. Both eosinophilia and HES can precede lymphoma’s development and are currently considered poor prognostic factors, according to the available data(1,2).
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Vlădăreanu AM. Actualităţi în limfoamele maligne nonhodgkiniene. Editura Medicală Amaltea, Bucureşti, 2002.
-
Helbig G, Wieczorkiewicz A, Dziaczkowska-Suszek J, Majewski M, Kyrcz-Krzemien S. T-cell abnormalities are present at high frequencies in patients with hypereosinophilic syndrome. Haematologica. 2009 Sep;94(9):1236.
-
Klion AD. How I treat hypereosinophilic syndromes. Blood. 2015 Aug 27;126(9):1069-77.
-
Valent P. Pathogenesis, classification, and therapy of eosinophilia and eosinophil disorders. Blood Reviews. 2009 Jul 1;23(4):157-65.