The primary central nervous system lymphoma (PCNSL) is a rare form of extranodal lymphoma that can affect the brain, the leptomeninges, the eye or the spinal cord, without any evidence of a systemic disease, having immunodeficiency as the main risk factor. The clinical presentation varies depending on the affected area. The diagnosis starts with a clinical suspicion, and must be supplemented by laboratory tests (blood and cerebrospinal fluid analysis) and imaging examinations. Even though the therapeutic protocols are limited, using the new treatments significantly increased the overall survival during time, but tumor relapse is still frequent and generally with poor prognosis.
HEMATOLOGY
Particularităţile diagnosticului în limfoamele primare ale sistemului nervos central (PCNSL)
Peculiarities of diagnosis in primary central nervous system lymphomas (PCNSL)
Anca Nicolescu,
Ana Maria Prof. Dr. Vlădăreanu,
Conf. Dr. Horia Bumbea,
Diana Cisleanu,
Raluca Nistor,
Ion Dumitru,
Tiberiu Sultan
First published: 29 decembrie 2018
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/OnHe.45.4.2018.2171
Abstract
Rezumat
Limfomul primar al sistemului nervos central este o formă rară de limfom extranodal care poate afecta creierul, leptomeningele şi ochiul sau măduva spinării, fără a exista dovezi ale unei boli sistemice, având imunodeficienţa ca principal factor de risc. Prezentarea clinică diferă în funcţie de zona afectată. Diagnosticul porneşte de la suspiciunea clinică şi trebuie completat de teste de laborator (de sânge şi ale lichidului cefalorahidian) şi de examinări imagistice. Deşi protocoalele terapeutice sunt limitate, noile tratamente disponibile au dus la îmbunătăţirea semnificativă a supravieţuirii globale, însă recidivele tumorale sunt frecvente şi în general cu prognostic slab.
The primary central nervous system lymphomas (PCNSL) are rare tumors (3% of the brain tumors), representing roughly about 1-3% of the non-Hodgkin’s lymphomas (NHLs) and 2-3% of the extranodal NHLs. The incidence is 4 cases/million/year. For the immunocompetent patients these are extremely rare tumors, typically for them being a single, solitary tumor; the incidence is much higher in the immunosuppressed patients, when PCNSLs occur in the form of multiple tumoral formations. Immunodeficiency (HIV infection, iatrogenic immunosuppression and congenital immune disorders) is the main risk factor(1,2) in the development of PCNSL, but PCNSL may also occur sporadically in individuals apparently immunocompetent in association with other conditions (frequently some viral or gastrointestinal infections, autoimmune diseases). PCNSL typically affects elderly patients (the median age at the diagnosis is 66 years old)(3,4).
PCNSL may associate manifestations in the brain, the meninges, the eye, the spinal cord, five distinct clinical-pathological entities being described. The symptoms and the clinical signs vary depending on the site of affection(5,6,7):
1. Intracranial lesions (solitary or multiple)
2. Diffuse or periventricular leptomeningeal lesions
3. Uveal and vitreous lymphoma deposits
4. Intradural lesions in the spinal cord
5. Neurolymphomatosis.
1. The central nervous system lymphoma. The clinical presentation includes suggestive symptoms for a periventricular lesion in the brain: focal neurologic deficits (70%), neuropsychiatric symptoms (43%), increased intracranial pressure syndrome (33%), epileptic seizures (14%), ocular symptoms (4%)(8). The onset symptoms may also include cephalalgia, visual disturbances, motor impairment and personality disorders such as depression, apathy, confusion, psychosis, memory impairment and hallucinations. The personality disorders are commonly associated with lesions of the frontal lobe or of the corpus callosum. The visual hallucinations may be due to infiltration of the visual pathways, of the brain stem, of the ocular or leptomeninges invasion. Facial nerve palsy may occur in the context of meningeal damage, brain stem infiltration, or isolated damage of the cranial nerves or nerve roots. Cephalalgia may be an indication of increased intracranial pressure or invasion of the leptomeninges.
2. The primary leptomeningeal lymphoma (PLML). More than 40% of PCNSL patients may have meningeal impairment at their diagnosis, diagnosed by cerebrospinal fluid (CSF) or neuroimaging analysis; also, the leptomeningeal impairment is a frequent pattern in the case of high-risk systemic lymphomas relapse. The patients with primary leptomeningeal lymphoma have signs and symptoms of neuraxial injury that may include severe cephalalgia, cranial nerve damage, meningitis, cervical or lumbar radiculopathies, ataxia and encephalopathy. The CSF analysis is abnormal in all cases. The cytological examination of the CSF reveals the presence of malignant lymphocytes. The monoclonal population can be detected by flow cytometry in 80% of the cases.
3. The primary intraocular lymphoma (PIOL) is a form of PCNSL with initial ocular manifestation with or without CNS damage. The diagnosis can be determined by biopsy of the vitreous body, of the choroid or of the retina. The primary intraocular lymphoma should be differentiated from the retro-orbital lymphoma that is frequently associated with the extranodal systemic disease. Ocular damage as an initial manifestation of the PCNSL may be unilateral or bilateral, frequently preceding the manifestations associated with the lymphoma infiltration in the CNS parenchyma. The vitreous body and the uvea may frequently be relapsing sites of a cerebral lymphoma or, rarer, of a systemic lymphoma(9,10).The eye symptoms are often subtle and nonspecific, and cannot be easily distinguished from those due to sarcoidosis, viral infections or collagen vascular diseases(11,12,13). The patients diagnosed with ocular localization need to undergo brain MRI and lumbar puncture with cytological examination of the CSF(14).
4. The spinal primary lymphoma represents less than 1% of all diagnosed PCNSLs; it manifests with discrete intramedullary nodal lesions, unlike the spinal localizations of the systemic lymphomas where there is usually a diffuse leptomeningeal impairment or one with extracorporeal nodules. Still, in both cases, the spinal localizations are manifested in the form of myelopathy, with a different clinical presentation depending on the site and extent of the lesion. Most patients experience progressive myelopathy, B symptoms (fever, night sweats, weight loss, altered general state), lumbar pain, areflexia or flaccid paralysis.
5. Neurolymphomatosis is the invasion of the cranial or spinal nerve roots with the following symptomatology: hypoesthesia, impaired motor function at the facial level and asymmetric weakness of the extremities(15,16). Optic nerve infiltration cases have also been described in the literature with sudden and severe loss of the visual function(17). The main feature of neurolymphomatosis is severe pain with imprecise localization. The meninges are often unaffected.
The PCNSL diagnosis usually starts from a clinical suspicion following a complete neurological examination, considering the wide and inconsistent spectrum of the symptomatology that can be missed by the diagnosis for several months. Subsequently, additional investigations, such as CSF analysis (cytology, biochemistry, bacteriological tests, flow cytometry, PCR, biomarkers analysis) are needed, followed by imaging investigations: MRI with contrast (to be preferred), CT scan (the most frequently used), PET-CT, and PET-MRI. If after performing all these investigations the diagnosis remains inconclusive, the stereotactic biopsy with the histopathological, immunohistochemical and immunophenotyping examination of the biopsy specimen remains the gold standard technique for the diagnosis of PCNSL, but in many cases this cannot be done because of patient-related reasons or tumor localization.
a. CSF Citology. This examination can provide diagnostic information in PCNSL by morphologically identifying the tumor cells in the CSF in patients with leptomeningeal localization. The method has 100% specificity and only 50% sensitivity at the first lumbar puncture. The results may be false positive for sample contamination and false negative; the absence of tumor cells does not rule out a possible CNS damage.
b. The biochemical analysis of the CSF usually reveals moderate proteinorrhachia (rarely above 5 g/l), hypoglycorrhachia, moderate pleiocytosis with predominant lymphocytes and mononuclear cells, as well as tumor cell presence. Beta-2 microglobulin can be elevated in the CSF. The method presents a difficulty in the morphological differentiation between the neoplastic cells and the inflammatory lymphocytes; in this regard, it is necessary to carry out immunohistochemical tests or cell immunophenotyping in order to identify the lymphoid cells with atypical, monoclonal or neoplastic morphology.
c. The cytometric flow pattern of the CSF allows the detection of the malignant cells by immunophenotyping with the lymphoid cell immunophenotype determination by CSF analysis or cell suspension analysis obtained after the tissue dissociation of the biopsy specimen. Immunophenotyping is also recommended in patients with aggressive NHLs at risk of CNS impairment. More than 80% of LNH cases with brain involvement can be diagnosed at the first flow cytometry of the CSF. The method has a very high sensitivity compared to the cytology examination and a high specificity in the clonal lymphoid cell detection, having a diagnostic role for the CNS impairment in the patients with lymphoma with leptomeningeal localization. There may be false positive results if the CSF is contaminated with lymphocytes from the peripheral blood during the lumbar puncture, or false negative results, knowing the fact that the CSF cells are fragile and require harvesting in stable suspensions and rapid screening. The PCNSL immunophenotype is most of the times (>95% cases) of the diffuse large B-cell lymphomas: CD45+, CD19+, CD20+, CD22+, CD79b+/-, CD5-, CD10+/-, CD34-; +/- T cell infiltration: CD2, CD3, CD5, CD8+. The level of soluble CD27 (GP transmembrane expressed by most mature T cells and B cells with memory) shows diagnostic utility in the NHLs with leptomeningeal localization.
d. The biomarkers that can be identified by CSF analysis are: IL10 (decreases after treatment, increases in relapses); IL6 (exhibits significantly elevated level in PCNSL compared to other brain tumors), osteopontin (proinflammatory CK, presents high level in PCNSL compared to the inflammatory diseases and gliomas), sCD19, AT III, light chains Kappa, lambda, CXCL13, miRNA. Biomarkers play a key role in determining the prognosis of PCNSL.
e. The stereotactic biopsy remains the procedure of choice in the diagnosis of PCNSL if there is no ocular or meningeal damage. The extensive resection of the lesion cannot be practiced in most of the cases due to its profound localization. Usually, the use of corticosteroids prior to biopsy is avoided because of the lymphocytotoxic effect – a single administration may alter the histopathological analysis, whereas a short-term treatment could lead to the temporary disappearance of the tumor.
It is recommended that the histopathological evaluation include both the microscopic examination and the immunohistochemistry. Occasionally, the first examination may be inconclusive, there may be suggestive sentinel lesions for demyelination or viral encephalitis that will later on develop into a lymphoma(5,11).
The method has reduced complications, especially if performed under the MR guideline. The PCNSL histological subtypes are: 96% aggressive NHLs (most commonly DLBCLs) and 4% indolent NHLs, and are predominantly with B-cell (98%), rarely with T cell. Immunohistochemically, PCNSLs are frequently positive for BCL6, MUM1 and BCL2, and negative for CD138-, CD38-, and plasma differentiation markers(18).
Histopathologically, the tumor cells are large centroblastic or immunoblastic cells that tend to invade the perivascular spaces, the adjacent parenchyma as well as the vascular wall; there also may be present reactive astrocytes, macrophages, and rarely infiltration with T cells.
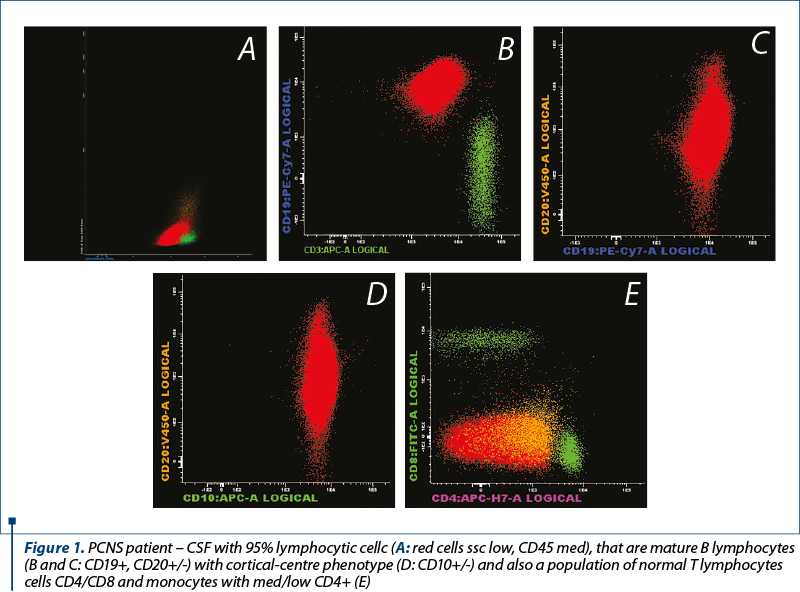
f. Imaging investigations – CT scan
CT scan is the most commonly used imaging investigation. The characteristic of PCNSL are the hyperdense, homogenous lesions, located in the periventricular white substance, in the corpus callosum or more rarely in the basal ganglia. The lesions are frequently single (70%) or multiple/multifocal (30%) in the immunocompromised patients.
g. MRI with contrast medium is the imaging technique of choice for the diagnosis of PCNSL. MRI plays an important role in guiding the biopsy procedures, performing the therapeutic plan for radiation therapy, or establishing the surgical decompression treatment for the tumors with surgical indication. The characteristic of PCNSL are the masses with homogeneous contrast medium contact with clearly defined edges, frequent perilesional edema, homogeneous appearance in T1, and T2-isofunctional or hypofunctional lesions with restricted diffusion(18).
Also, MRI has an important role in the differential diagnosis. In HGG (high grade glioma) the MRI aspect is of heterogeneous lesions with necrotic areas. The demyelinating lesions are usually solitary, T2-hyperintense, larger than 2 cm, with mass effect and edema. In the case of infections, such as the cerebral toxoplasmosis and of granulomatous diseases (neurosarcoidosis), the onset is acute, and on the MRI there may be present T1-iso-/hypointense lesions and T2-hyper-/hypo-/isointense (depending on the abscess stage) localized at the level of the basal ganglia. The secondary cerebral localizations are frequently found in the gray matter, at the level of the cerebral hemispheres, cerebellum and basal ganglia, and may be single/multiple, with central necrosis, ring-enhancing lesion and edema(18). In order to complete the differential diagnosis, cervical-thoracic-abdominal-pelvic CTs may be required, as well as osteomedullary biopsy, testicular ultrasound, bacteriological, virological and immunological tests(19).
h. PET combined with CT/MRI brings detailed information on cellular metabolism, highlighting the FDG capture area and with a role in guiding the stereotactic biopsy. The PET-CT differentiates between low or increased malignancy tumors (especially gliomas) and has a major impact on the surgical decision in the pediatric patients with brain tumors (precise delineation of the lesion). The PET-CT can highlight the early tumor lesions with an important role in diagnosis and staging, having a high sensitivity for the diagnosis of the systemic disease in comparison to the CT scan. The PET-CT can highlight the remaining post-chemotherapy masses with a role in the subsequent therapeutic decision. In the event of suspicion of relapse or progression of the disease, the PET-CT may reveal new tumor masses. Also, the post-irradiation tumor necrosis is differentiated from a residual tumor mass by the PET-CT. For patients who cannot undergo a cerebral biopsy, a therapeutic test can be performed to detect the subcortical, intense, homogenous FDG capture at the level of the tumor before corticotherapy and the reducing in the tumor size (as evidenced by the MRI) after one week of cortisone treatment. Last but not least, the PET-CT contributes to the differential diagnosis of cerebral tumors with infections (especially with cerebral toxoplasmosis) and the granulomatous diseases(19).
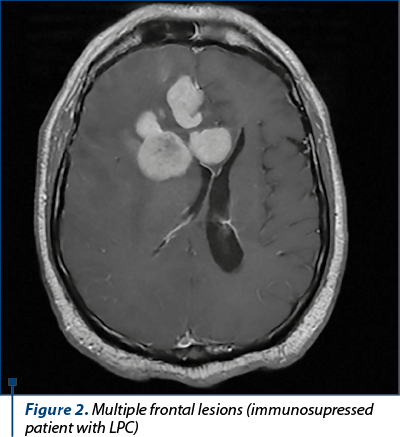
PET-MRI, the more recently developed technique compared to the PET-CT, has as advantages a greater accuracy in detecting the tumor lesions in the brain, breast, liver, kidneys and bone, and the exposure to radiation by 50%, depending on the protocol(19).
Analyzing the techniques used in the diagnosis of PCNSL, Brian J. Scott et al. demonstrated by successive studies that the most sensitive techniques are: CT/MRI with contrast medium (99%), cerebral biopsy and micro-RNA analysis (96%); and the techniques with the highest specificity are: ATIII level (98%), micro-RNA analysis (97%), CSF cytology (100%) and cerebral biopsy (100%)(20).
The same team of researchers proposed a diagnostic algorithm for PCNSL. After performing a complete neurological examination (monitoring the motor deficits, the psychiatric symptoms, the presence of HIT signs, the ocular symptoms), they proposed to initiate the investigations with routine serological tests (complete blood count, biochemistry, blood coagulation test, HIV test), followed by brain MRI exam with i.v. contrast medium. Spinal MRI will be performed only if there is specific symptomatology (myelopathy). The characteristics of PCNSL are the T1-hypointense MRI and T2-iso- or hypointense lesions, homogeneous and with restricted diffusion, typically located in the cerebral hemispheres, thalamus, corpus callosum, cerebellum, eye, meninges or spinal cord. It is recommended to perform the ophthalmic examination with a slit lamp to exclude an ocular localization. Lumbar puncture is then recommended to be performed together with some CSF tests: cytology, flow cytometry, biochemistry, biomarker analysis, PCR, micro-RNA analysis, EBV by PCR. To exclude a systemic disease and for staging, a thoracic-abdominal-pelvic CT scan and a testicular echography are indicated. If eye or ganglion damage is detected, a vitreous biopsy, respectively a ganglionic one, is indicated. If after these investigations the diagnosis remains uncertain, a stereotactic biopsy is indicated, preferably a MRI-guided one. After the diagnosis of PCNSL has been confirmed, staging, toxicity evaluation (biochemistry, echocardiography, respiratory function samples) and prognostic factors evaluation are performed. The IELSG prognostic score for PCNSL takes into account the following factors: age>60 years old, ECOG>2, elevated LDH, increase of CRL proteins and the presence of deep brain lesions(20).
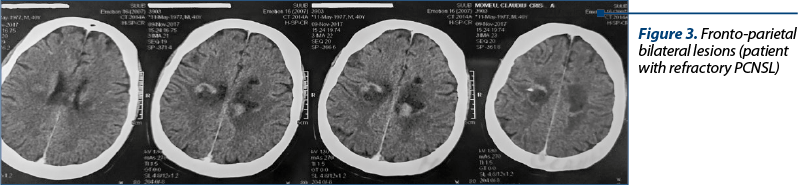
The treatment is performed according to the degree and type of CNS localization. The surgical treatment and the tumor excision do not bring benefits; the PFS may increase if the lesion is unique and superficial. Corticotherapy can delay the diagnosis, the PCNSL being very sensitive to corticotherapy. Typically, it has rapid radiologic regression under corticotherapy (40%) – which can create a diagnostic confusion with the inflammatory/demyelinating lesions. Corticotherapy is frequently associated with the therapeutic regimens due to the anti-edematous effect and a direct lymphocytolytic effect. The intrathecal chemotherapy plays an important role, having a direct CNS action, being included in most chemotherapy regimens. Out of the systemic chemotherapy drugs, only some medicines pass the blood-brain barrier. Almost all of the schemes contain methotrexate (3.5-8 g/m2) and cytarabine (Cytosar®), usually in high doses, to allow penetration into the CSF. PCNSLs are very susceptible to RT, but WBRT, as a treatment-enhancing therapy with methotrexate, has no benefit on the global survival.
With current treatments, survival has increased significantly over time, the current studies showing a two-year survival between 42% and 81% and a three-year survival between 32% and 46%, the tumor recurrence being still common and generally with uncertain prognosis.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
- Schabet M. Epidemiology of primary CNS lymphoma. J Neurooncol. 1999; 43:199.
- Bhagavathi S, Wilson JD. Primary central nervous system lymphoma. Arch Pathol Lab Med. 2008; 132:1830.
- Hoffman S, Propp JM, McCarthy BJ. Temporal trends in incidence of primary brain tumors in the United States, 1985-1999. Neuro Oncol. 2006; 8:27.
- Villano JL, Koshy M, Shaikh H, et al. Age, gender, and racial differences in incidence and survival in primary CNS lymphoma. Br J Cancer. 2011; 105:1414.
- Hochberg FH, Miller DC. Primary central nervous system lymphoma. J Neurosurg. 1988; 68:835.
- Fine HA, Mayer RJ. Primary central nervous system lymphoma. Ann Intern Med. 1993; 119:1093.
- Herrlinger U, Schabet M, Clemens M, et al. Clinical presentation and therapeutic outcome in 26 patients with primary CNS lymphoma. Acta Neurol Scand. 1998; 97:257.
- Bataille B, Delwail V, Menet E, et al. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg. 2000; 92:261.
- Salomão DR, Pulido JS, Johnston PB, et al. Vitreoretinal presentation of secondary large B-cell lymphoma in patients with systemic lymphoma. JAMA Ophthalmol. 2013; 131:1151.
- Mashayekhi A, Shukla SY, Shields JA, Shields CL. Choroidal lymphoma: clinical features and association with systemic lymphoma. Ophthalmology. 2014; 121:342.
- Rock JP, Cher L, Hochberg FH, et al. Central nervous system lymphomas in AIDS and non-AIDS patients. In: Neurological Surgery, 4th ed, Yomans JR (Ed), WB Saunders, Philadelphia, 1995; p. 593.
- Hormigo A, Abrey L, Heinemann MH, DeAngelis LM. Ocular presentation of primary central nervous system lymphoma: diagnosis and treatment. Br J Haematol. 2004; 126:202.
- Rivero ME, Kuppermann BD, Wiley CA, et al. Acquired immunodeficiency syndrome-related intraocular B-cell lymphoma. Arch Ophthalmol. 1999; 117:616.
- Grimm SA, McCannel CA, Omuro AM, et al. Primary CNS lymphoma with intraocular involvement: International PCNSL Collaborative Group Report. Neurology. 2008; 71:1355.
- Khong P, Pitham T, Owler B. Isolated neurolymphomatosis of the cauda equina and filum terminale: case report. Spine (Phila Pa 1976). 2008; 33:E807.
- Da Silva AN, Lopes MB, Schiff D. Rare pathological variants and presentations of primary central nervous system lymphomas. Neurosurg Focus. 2006; 21:E7.
- Ahle G, Touitou V, Cassoux N, et al. Optic Nerve Infiltration in Primary Central Nervous System Lymphoma. JAMA Neurol. 2017; 74:1368.
- Braaten KM, Betensky RA, de Leval L, et al. BCL-6 expression predicts improved survival in patients with primary central nervous system lymphoma. Clin Cancer Res. 2003; 9:1063.
- Rosenkrantz AB, Friedman K, Chandarana H. Current Status of Hybrid PET/MRI in Oncologic Imaging. Am J Roentgenol. 2016 Jan; 206(1): 162–172.
- Chiavazza C, Pellerino A, Ferrio F, Cistaro A, Soffietti R, Rudà R. Primary CNS Lymphomas: Challenges in Diagnosis and Monitoring. Biomed Res Int. 2018 Jun 21; 2018:3606970.
- Scott BJ, Douglas VT et al. A Systematic Approach to the Diagnosis of Suspected Central Nervous System Lymphoma. JAMA Neurol. 2013 Mar 1; 70(3): 311–319.
Articole din ediţiile anterioare
ORIGINAL STUDY | Ediţia 2 51 / 2020
Importanţa evaluării computer-tomografice în detecţia interesării tumorale limfomatoase cardio-pericardice
Anca Flintoacă-Filip, Adriana Badea- Selea, Ioana G. Lupescu
Leucemiile şi limfoamele maligne interesează frecvent structurile cardio-pericardice, cel mai adesea prin extensie directă de la o masă medias...
19 mai 2020
HEMATO-ONCOLOGY | Ediţia 1 46 / 2019
Un caz complex de limfom Hodgkin clasic recidivat refractar – scleroză nodulară
Alina Mititelu, Elena Andruş-Lupoaia, Șef de lucrări dr. Minodora Onisâi, Andreea Spînu, Camelia Dobre, Alina Tănase, Dragoş Bumbăcea, Ana Maria Prof. Dr. Vlădăreanu
Raportăm cazul unui pacient de 32 de ani care s-a prezentat în departamentul nostru în mai 2016 cu simptome respiratorii (tuse, dispnee) şi febră. ...
27 martie 2019
ONCOHEMATOLOGY | Ediţia 1 58 / 2022
A rare case of highly aggressive Burkitt lymphoma with multiple extranodal involvements of the elderly
Iuliana Iordan, Andreea Neculcea, Diana Emanuela Bonea, Dragoş-Claudiu Popescu, Alina Mititelu, Andreea Spînu, Paul Findrihan, Raluca Nistor, Ana Maria Prof. Dr. Vlădăreanu
Limfomul Burkitt este unul dintre cele mai agresive limfoame non-Hodgkin. Poate fi însoţit de determinări extranodale, cele mai frecvente fiind l...
25 martie 2022
HEMATO-ONCOLOGY | Ediţia 1 66 / 2024
Images in hematology: adult T-cell leukemia/lymphoma
Iuliana Iordan, Ana Maria Neagu, Cristina Mambet, Mihai Stejara, Ana Maria Vlădăreanu
Leucemia/limfomul cu celule T al adultului (ATLL) este un neoplasm agresiv al celulelor T, cauzat de infecţia cu virusul limfotrop de tip 1 al celu...
27 martie 2024