Hyper-IgE syndrome – from pathogenic complexity to clinical manifestations
Sindromul hiper-IgE – de la complexitate patogenică la manifestări clinice
Abstract
Hyperimmunoglobulin E syndrome (HIES) is a rare primary immunodeficiency characterized by increased serum IgE levels associated with eczema, recurrent lung and skin infections and with various connective tissue, skeletal and vascular abnormalities. Disease mechanisms involve STAT3 signalling pathway, deficiency of which leads to synthesis of many T helper 1 (Th1) cytokines, such as IFN-γ and TNF-α, and inhibition of proinflammatory and antiinflammatory responses regulated by IL-6 and IL-10. Clinical features of HIES involve the immune system, connective tissue, skeleton, skin and dentition, with variations in severity and timing of onset. The early recognition of HIES can lead to a better treatment of the associated complications.Keywords
hyperimmunoglobulin E syndromeimmunodefficiencyRezumat
Sindromul hiperimunoglobulinemiei E (HIES) este o imunodeficienţă primară rară caracterizată prin niveluri serice crescute de IgE asociate cu eczeme, infecţii pulmonare şi cutanate recurente şi cu diverse anomalii ale ţesutului conjunctiv, scheletului şi vaselor. Mecanismele bolii implică o cale de semnalizare STAT3, a cărei deficienţă duce la sinteza multor citokine T helper 1 (IFN-γ şi TNF-α) şi la inhibarea răspunsurilor proinflamatorii şi antiinflamatorii reglate de IL-6 şi IL-10. Caracteristicile clinice ale HIES implică sistemul imunitar, ţesutul conjunctiv, scheletul, pielea şi dentiţia, cu variaţii în ceea ce priveşte severitatea şi momentul de apariţie. Recunoaşterea timpurie a HIES poate duce la o mai bună tratare precoce a complicaţiilor asociate.Cuvinte Cheie
sindromul hiperimunoglobulinemiei EimunodeficienţăBackground
Atopy is the individual’s genetic predisposition to develop specific, highly heterogeneous responses mediated mostly or exclusively by IgE antibodies to common environmental antigens.
Immunodeficiency is the functional failure or absence of elements of the immune system, including lymphocytes, phagocytes and the complement system. It may be of primary (congenital) cause or it may be secondary(15).
Hyperimmunoglobulin E syndrome, or HIES, is a rare primary immunodeficiency characterized by eczema, recurrent lung and skin infections, increased serum IgE levels and various connective tissue, skeletal and vascular abnormalities(11).
Epidemiology
Hyperimmunoglobulin E syndrome is rare. The exact incidence is not known and is estimated to range from 1 in 500,000 to 1 in 1,000,000 people. It occurs equally in both sexes. It has been reported in Caucasians as well as in individuals of Asian and African descent(18).
This syndrome was first reported in 1966 by Davis et al., who presented the cases of two red-haired Caucasian girls with frequent staphylococcal skin and sinopulmonary infections, chronic dermatitis and an abnormal inflammatory response. They named the condition Job’s syndrome, based on its similarity to the illness of the biblical character, whose body was covered with painful boils. Elevated IgE levels and a neutrophil chemotaxis defect were identified in the two girls with Job syndrome, showing that hyper-IgE syndrome and Job syndrome are probably the same disorder(4).
In 1972, Buckley and co-workers brought two boys with similar problems to their attention, namely severe dermatitis, recurrent cutaneous, pulmonary and joint abscesses, and coarse facies and growth retardation, with elevated serum IgE levels and eosinophilia. This presentation has been termed Buckley syndrome(3).
These two syndromes were later found to fall into the same category and were introduced under the new title of HIES. HIES was also well described by Hill and Quie in 1974, by Donabedian and Gallin in 1983, and by Belohradsky and co-workers in 1987. A systemic evaluation of thirty patients determined that HIES is a multisystem disorder characterized by susceptibility to infection, elevated serum IgE levels, eosinophilia, distinctive facial appearance by age 16, preserved primary dentition, bone fragility, hyperextensible joints, scoliosis and craniosynostosis(8).
Pathogenic mechanisms
HIES is classified into two types.
-
Type I: autosomal dominant hyper-IgE syndrome (AD-HIES), in which patients have abnormalities in various systems, including the immune system, connective tissue, vessels and skeleton.
-
Type II: autosomal recessive hyper-IgE syndrome (AR-HIES), also affecting the immune system, manifested by elevated serum immunoglobulin E values, recurrent skin and lung infections, susceptibility to viral infections such as Molluscum contagiosum and central nervous system involvement, but without musculoskeletal changes(9).
The majority of AD-HIES patients have a defect in the signal transducer and activator of transcription 3 (STAT3), which is encoded on chromosome 17q21(1). Most of the reported mutations are single amino acid nonsense, but amino acid deletions or exon omissions have also been reported. The changes are concentrated in the SH2 domain (involved in mediating protein-protein interactions) and the DNA-binding domain (involved in mediating protein-DNA interactions) of STAT3(12).
STAT3 is one of six STAT proteins that are major signal transducers involved in multiple signaling pathways(16). It is a cytoplasmic protein and a component of the JAK-STAT (Janus kinase) signal transduction pathway initiated by a multitude of cytokines, growth factors and hormones, resulting in multisystemic damage in patients suffering from hyperimmunoglobulin E syndrome. Intracellular components of cytokine receptors bind to one of four Jak proteins (Jak1, Jak2, Jak3, Tyk2), leading to STAT recruitment and phosphorylation. Phosphorylated STAT molecules then dimerize at the SH2 domain and are translocated to the nucleus, leading to the activation of STAT-regulated genes (Figure 1).
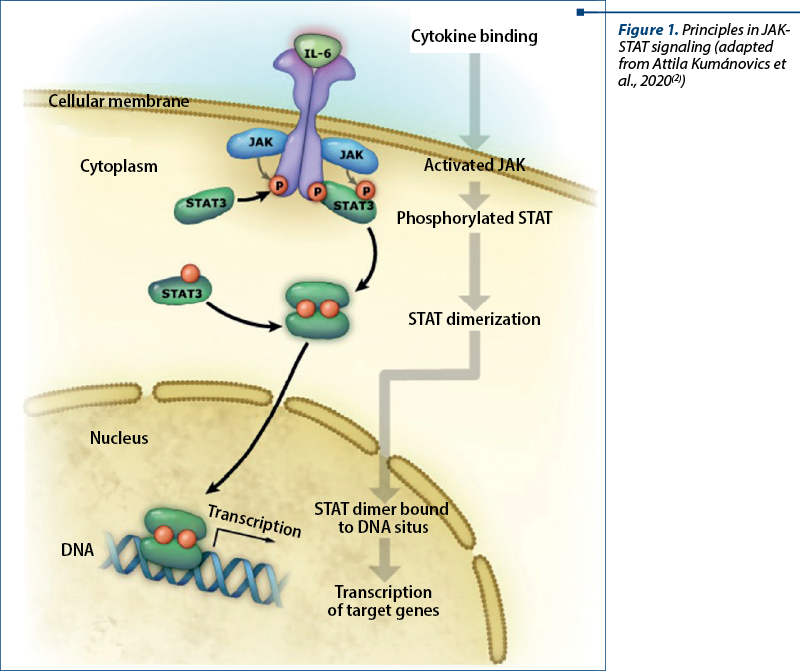
STAT3 is involved in signal transduction of many cytokines, including but not limited to interleukin 6 (IL-6), interleukin 10 (IL-10), interleukin 17A/F (IL-17A/F), interleukin 22 (IL-22), interleukin 26 (IL-26), and plays an important role in wound healing, angiogenesis, cancer and immunity. On the other hand, proinflammatory mediators, including tumor necrosis factor a (TNF-a), interleukin 12 (IL-12) and interferon gamma (IFN-g), are inhibited by STAT3 signaling. In normal cells, STAT3 activation is a transient and well-controlled process, after which dephosphorylated STAT3 is transferred back into the cytoplasm through the nuclear pores(18).
Therefore, STAT3 deficiency leads to the synthesis of many T helper 1 (Th1) cytokines, such as IFN-g and TNF-a, and to the inhibition of proinflammatory and antiinflammatory responses regulated by IL-6 and IL-10(12).
STAT3 plays a central role in the IL-6 signaling pathway and in the induction of the acute phase response, and therefore impaired IL-6 function is thought to be responsible for a large part of the immunodeficiency present in HIES. In addition, IL-22, acting through STAT3, has been found to play a role in regulating epithelial cell barrier function which is also disrupted in HIES(18).
After receptor binding of a relevant cytokine or growth factor, the receptor undergoes homo- or heterodimerization and binds cytosolic JAKs (JAK1, 2, 3 or Tyk2) for autophosphorylation and receptor transactivation. This event allows the recruitment of transcription factors belonging to the STAT family (STAT 1, 2, 3, 4, 5A, 5B or 6) which bind the cytoplasmic domain of the receptor through their SH2 domain. Phosphorylated STAT proteins subsequently undergo homo- or heterodimerization and translocate to the nucleus, where they induce transcriptional activation of target genes (Figure 1).
The occurrence of a significantly decreased T helper 17 (Th17) cell response is a hallmark of HIES, with STAT3 mutations shown to lead to a failure of Th17 CD4 cell differentiation (differentiation cluster) – Figure 2. IL-17 synthesis by Th17 cells is involved in chemotaxis and neutrophil proliferation. Thus, the defective neutrophil response to skin and lung infections may be responsible for recurrent infections with these sites. Respiratory epithelial cells and keratinocytes are dependent on IL-17 to produce antimicrobial peptides, including those for antifungal responses. Abnormal production of this interleukin is probably also involved in the interferon gamma (IFN-g) synthesis pathway in AD-HIES patients, as their mononuclear cells secrete low amounts of IFN-g when stimulated(17).
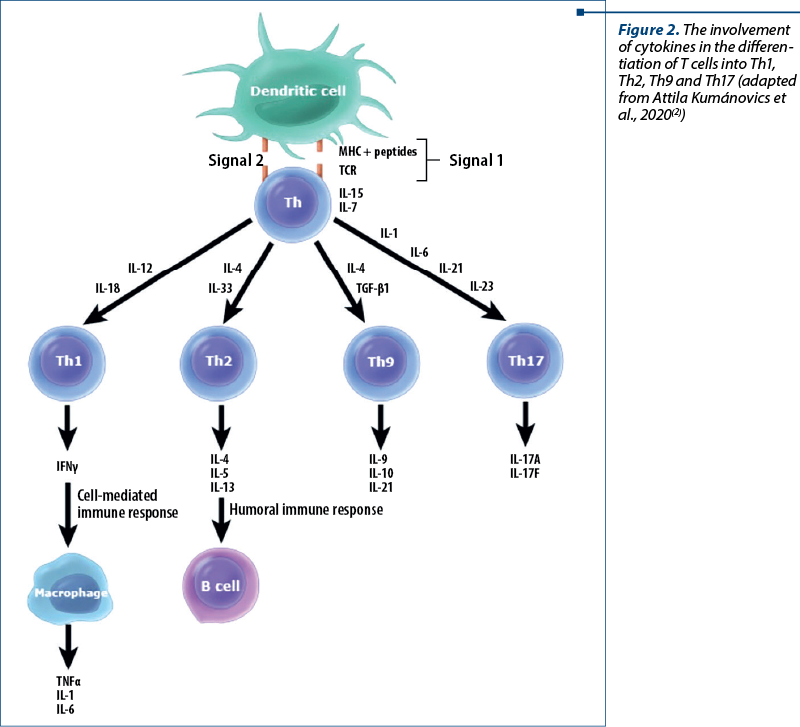
STAT3 is also involved in the differentiation of T helper 9 (Th9) cells (Figure 2). Th9 lymphocytes produce IL-9, among other things, and plays a role in allergy, asthma and autoimmune inflammation, have antitumor, antihelminthic and antiprotozoal effects, and are involved in other extracellular pathogenic responses.
In 2009, heterozygous and homozygous mutations present in the Dedicated Cytokinase 8 (DOCK8) gene were identified in patients previously described as having autosomal recessive hyperimmunoglobulin E syndrome (AR-HIES). The presence of these mutations is responsible for the lack of DOCK8 expression. Several clinical features differentiate DOCK8-deficient patients from those with AD-HIES, including a lack of non-immunological features and a higher incidence of viral skin infections and malignancies(20).
DOCK8 is involved in organizing the actin cytoskeleton for cell migration and synapse formation. Lymphocytes that are deficient in DOCK8 exhibit disruptions in migration through the collagen matrix, this being incriminated to be one of the factors in the high incidence of severe cutaneous viral infections. DOCK8 is also involved in the survival of T, B and natural killer (NK) lymphocytes(5).
Also, heterozygous and homozygous mutations in phosphoglucomutase 3 (PGM3) were identified underlying a distinct phenotype of hyper-IgE syndrome in 2014(14). PGM3 is involved at an early stage in many glycosylation pathways, being responsible for the conversion of glucosyl-N-acetyl-6-phosphate to glucosyl-N-acetyl-1-phosphate, which is then converted to uridine diphosphate N-acetylglucosamine (UDP-GlcNac). Glycosylation is necessary for the proper functioning of most proteins. The pathogenesis of the multiple features of this disease is still under investigation(19).
IgE-mediated immune response in atopic individuals
The genetic alteration of the barrier function of the skin and/or gastrointestinal mucosa, immature in infants and young children or affected by various inflammatory processes, increases antigen exposure, causing loss of hyporesponsiveness of dendritic and epithelial cells expressing molecules involved in the differentiation of naive Th lymphocytes into Th2 or IgE-secreting plasma cells.
The IgE-mediated allergic immune response is a three-step process. The sensitization stage begins with uptake and processing of antigen by antigen-presenting cells and the subsequent presentation to naive CD4+ T lymphocytes. The cytokines IL-4 and IL-13 are involved in the differentiation of Th0 lymphocytes into Th2 lymphocytes, with a role in the phenotypic transformation of B lymphocytes into IgE-secreting plasma cells. The immunoglobulin formed binds via high affinity receptors (FceRI) to the surface of mast cells and basophils.
The subsequent exposure to the same antigen will be followed by its cross-binding to two IgE molecules and followed by the activation of basophil and mast cells, resulting in the release of preformed mediators from intracytoplasmic granules. The acute reaction occurs in the first seconds to minutes and may be followed by a late reaction, characterized by tissue infiltration with granulocytes and lymphocytes, Th2 in particular, after 2 to 24 hours.
The chronic phase, presumed to be the result of perpetuation of late reactions, is not Th2-dominated and is characterized by stimulation of sensory nerves, increased vascular permeability, arteriolar dilation and impaired gastrointestinal function, resulting in structural changes such as fibrosis and organ dysfunction.
Clinical picture – common elements and individual particularities
The clinical features of HIES involve the immune system, connective tissue, skeleton, skin and dentition, with variations in severity and timing of onset(10). HIES manifests as dermatitis and recurrent infections (mainly bacterial lung and skin infections), although there are significant variations in the constellation of symptoms and signs in different patients. The frequency of food allergy and anaphylaxis is significantly lower in patients with HIES compared to patients with atopic dermatitis and similar IgE concentrations (food allergy 38% versus 58%, and food-induced anaphylaxis 8% versus 33%, respectively)(13).
The clinical manifestations reported in 85 patients entered into the United States Immunodeficiency Registry (USIDNET Registry) included:
-
Skin abscesses – 74%.
-
Eczema – 58%.
-
Other allergic manifestations – drug allergies (43%), food allergies (38%).
-
Persistence of primary dentition – 41%.
-
Fractures – 39%.
-
Scoliosis – 34%.
-
Neoplasia – 7%(7).
The first manifestation of the disease is usually a rash, which may be present at birth or appear in the first few weeks. It starts with pink or erythematous papules that may turn into pustules, discharge pus and become covered with scabs. Lichenification occurs either in a mild form or is absent altogether. The rash is usually diagnosed as acne neonatorum or erythema toxicum neonatorum and predominates on the face, scalp, neck and upper torso. Eosinophils may be evident in the cooling product. The lesions are intensely pruritic, the pruritus being attributed to the release of histamine from cutaneous mast cells in response to stimulation with staphylococcal antigens. The rash progresses to eczematous dermatitis through colonization and/or chronic infection with Staphylococcus aureus.
Skin abscesses or boils frequently occur in the early years of life and, similar to pyogenic pneumonia, are often associated with minimal systemic symptoms such as fever and pain. These abscesses may appear “cold” or devoid of classic signs and symptoms of inflammation, and pus is discharged on drainage, the most common microorganisms involved being Staphylococcus aureus and Candida albicans(6).
Lung infections are the second most common manifestation for AD-HIES patients, after skin manifestations. Patients often have chronic upper respiratory tract infections – chronic sinusitis, suppurative otitis media and mastoiditis, which may require surgery. The pathogens involved in these infections include S. aureus, C. albicans, Haemophilus influenzae, group A and B streptococci, along with Gram-negative pathogens such as Pseudomonas and fungi(2).
Recurrent pyogenic pneumonia caused by Staphylococcus aureus, Streptococcus pneumoniae and Haemophilus influenzae usually begins in the first years of life. Systemic signs of infection are often minor or absent, which explains the late diagnosis. For example, lobar pneumonia with S. aureus may present with minimal fever, normal peripheral white blood cell count and inflammatory markers close to normal values. Pyogenic pneumonia usually responds to appropriate antibiotics, but the late diagnosis is responsible for complications such as pleural empyema, pneumatocele and bronchiectasis. The altered epithelial repair process in AD-HIES explains the high frequency of pneumatocele and parenchymal abnormalities, found in about 70% of patients.
Once the lung parenchyma of AD-HIES patients has been altered by pyogenic pneumonia, the list of infecting pathogens expands to include nontuberculous mycobacteria (NTM), fungi such as Aspergillus fumigatus, and Gram-negative bacilli such as Pseudomonas aeruginosa. These chronic infections cause significant morbidity and mortality through catastrophic hemoptysis and multidrug resistance to antibiotics. P. carinii, Nocardia and intracellular mycobacteria are germs less commonly involved in pneumonia of patients with HIES-AD. Adult respiratory distress syndrome (ARDS) with disseminated intravascular coagulation (DIC) through methicillin-resistant S. aureus (MRSA) sepsis is a rare manifestation, but burdened by high mortality(6,10).
Of the opportunistic infections, the most common is cutaneomucous candidiasis with oropharyngeal, vaginal and/or ungual localization.
The gastrointestinal tract is also susceptible to infection. Cryptococcosis and colorectal histoplasmosis have similar symptoms to Crohn’s disease. Eosinophilic esophagitis and motility disorders, diverticulosis and, rarely, gastrointestinal perforations without clearly associated pathology have also been described.
Less common infections include disseminated candidiasis and disseminated or localized necrotizing mycobacterial infections after BCG immunization. Systemic candidiasis may be accompanied by endocarditis and localized tricuspid valve vegetations. Other opportunistic infections reported are cryptococcal meningitis and generalized lymphadenopathy caused by trichosporonosis.
One of the most distinctive markers in AR-HIES is refractory chronic viral infections, especially with herpes simplex virus (HSV), varicella-zoster virus (VZV) or Molluscum contagiosum (MC). HSV infection is manifested by recurrent aphthous lesions located around the nose and ear(10).
Medium-caliber artery abnormalities were most commonly recognized as coronary artery aneurysms and tortuosities, with increasing prevalence with age. Other vascular manifestations include cerebral aneurysm, which may present with subarachnoid hemorrhage, and pulmonary hemorrhage from dilated bronchial arteries adjacent to areas of chronic inflammation(6).
The ophthalmologic involvement includes conjunctivitis, keratitis, spontaneous corneal perforation, Candida or streptococcal endophthalmitis, recurrent giant chalazion, extensive xanthelasma, eyelid tumors, strabismus and bilateral keratoconus.
The distinct gross facial features of patients with HIES are defined by age 16 and include facial asymmetry with hemihypertrophy, prominent forehead, deep-set eyes, broad nasal base, broad nasal tip, increased interalar and interchal distance, mild prognathism and skin xerosis with prominent pores. Midline facial anomalies, such as cleft lip and palate, ogival palate and craniosynostosis, have been reported with a lower incidence, but are unusual.
Scoliosis is seen in 63% of patients and may be due to shortening of a limb, vertebral body abnormalities after thoracotomy or intrinsic susceptibility. Other skeletal abnormalities reported are osteopenia, joint hyperlaxity, various joint deformities, genu valgum, congenital spinal anomalies, as well as bifid rib, pseudoarthrosis of the rib and frequent pathological fractures.
HIES was also associated with a case of late osteogenesis imperfecta. The involvement of the primary and permanent dentition by persistent epithelial root sheath is a common manifestation of the disease. It is likely that delayed dental root resorption and ineffective inflammatory responses that result in pneumatocellular formation are both cytokine-mediated manifestations of osteoclast and macrophage activation.
Although autoimmune diseases are seen in all forms of HIES, especially AR-HIES, HIES has been associated with systemic lupus erythematosus (five cases documented in the literature), dermatomyositis, membranoproliferative glomerulonephritis, nephrotic syndrome, autoimmune thrombocytopenia (IPT), hemolytic anemia, bronchiolitis obliterans and with pericardial effusion.
Several malignancies associated with HIES have been described, suggesting that patients with HIES may be at an increased risk of neoplasia. Multiple cases of lymphomas have been reported, including Hodgkin lymphoma, non-Hodgkin lymphoma, Burkitt lymphoma and histiocytosis.
Lymphogenesis in HIES may be multifactorial, including chronic antigenic stimulation of polyclonal B lymphocytes by viruses, poor control of B cell proliferation of T cells, defective immunosurveillance and cytokine dysregulation. Many of the patients presented in the literature had advanced stage or extranodal disease, with a negative impact on vital prognosis.
Other cancers have been reported, including acute myeloid leukemia, vulvar and liver cancer, and metastatic squamous cell carcinoma of the tongue. However, lymphoma is the most common malignancy reported. A high index of suspicion for lymphoma should be considered in HIES patients presenting with adenopathy(10).
Conclusions
HIES remains a complex pathogenic entity, often underdiagnosed in children. Elevated serum IgE levels must always be connected with clinical and other biological findings in order to complete the picture of HIES. The early assessment of HIES can lead to a better prevention and treatment of the associated complications.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Alcántara-Montiel JC, Staines-Boone T, López-Herrera G, Berrón-Ruiz L, Borrego-Montoya CR, Santos-Argumedo L. Somatic mosaicism in B cells of a patient with autosomal dominant hyper IgE syndrome. Eur J Immunol. 2016;46(10):2438-2443.
-
Attila Kumánovics MT. UpToDate. Retrieved from UpToDate: https://www.uptodate.com/contents/autosomal-dominant-hyperimmunoglobulin-e-syndrome/print?search=hyper
-
Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49(1):59-70.
-
Davis SD, Schaller J, Wedgwood RJ. Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet. 1966;1(7445):1013-1015.
-
Zhang Q, Dove CG, Hor JL, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J Exp Med. 2014;211(13):2549-2566.
-
Freeman AF, Olivier KN. Hyper-IgE Syndromes and the Lung. Clin Chest Med. 2016;37(3):557-567.
-
Gernez Y, Freeman AF, Holland SM, et al. Autosomal Dominant Hyper-IgE Syndrome in the USIDNET Registry. J Allergy Clin Immunol Pract. 2018;6(3):996-1001.
-
Grimbacher B, Holland SM, Gallin JI, et al. Hyper-IgE syndrome with recurrent infections - an autosomal dominant multisystem disorder. N Engl J Med. 1999;340(9):692-702.
-
Hafsi W, et al. Job Syndrome. Stat Pearls. 2020 January.
-
Hashemi H, Mohebbi M, Mehravaran S, Mazloumi M, Jahanbani-Ardakani H, Abtahi SH. Hyperimmunoglobulin E syndrome: Genetics, immunopathogenesis, clinical findings, and treatment modalities. J Res Med Sci. 2017;22:53. Published 2017 Apr 26.
-
He YY, Liu B, Xiao XP. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2017;31(11):892-896.
-
Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1608-1619.
-
Ng C, Hon KL, Kung JS, Pong NH, Leung TF, Wong CK. Hyper IgE in Childhood Eczema and Risk of Asthma in Chinese Children. Molecules. 2016;21(6):753. doi:10.3390/molecules21060753.
-
Zhang Y, Yu X, Ichikawa M, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. 2014;133(5):1400-1409.e14095.
-
Justiz A. Immunodeficiency. Stat Pearls. 2020 Jan. Updated 2020 Aug 24.
-
Levy DE, Loomis CA. STAT3 signaling and the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1655-1658.
-
Moffitt K, Cheung E, Manis J, Malley R. Evaluation of the Role of stat3 in Antibody and TH17-Mediated Responses to Pneumococcal Immunization and Infection by Use of a Mouse Model of Autosomal Dominant Hyper-IgE Syndrome. Infect Immun. 2018;86(5):e00024-18.
-
Mogensen TH. STAT3 and the Hyper-IgE syndrome: Clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. JAKSTAT. 2013;2(2):e23435.
-
Stray-Pedersen A, Backe PH, Sorte HS, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. 2014;95(1):96-107.
-
Zhang Q, Davis JC, Lamborn IT, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361(21):2046-2055.

Diagnostic challenges in recurrent respiratory infections in children – the vascular ring. A case report
Bianca Raluca Mateescu, Alina Grama, Georgiana Laura Cioancă, Simona Căinap, Tudor Lucian Pop
Infecţiile respiratorii recurente sunt cauze frecvente de solicitare a consultului medical în populaţia pediatrică. Cauzele acestor manifestări sunt foarte variate, astfel încât o încadrare core...
Acute liver failure associated with recent SARS-CoV-2 infection in a pediatric patient – case report
Georgiana Laura Cioancă, Alina Grama, Luciana Petrescu, Simona Căinap, Bianca Raluca Mateescu, Gabriel Benţa, Bogdan Bulata, Tudor Lucian Pop
Insuficienţa hepatică acută (IHA) la copil este o patologie rară, dar severă, caracterizată prin coagulopatie şi modificări de lab...
Consequences of anemia in pregnancy upon the newborn – a cross-sectional study
Oana-Liliana Atomei, Paula-Paraschiva Monor, Bogdan A. Stana
Anemia la gravide este încă o problemă importantă de sănătate publică. Consecinţele asupra nou-născutului, imediate sau tardive, pot varia de la patologii uşoare la boli severe. Obiectivul principal al studiului...
Nutrition as a key factor in pediatric infectious disease
Iulia Ţincu, Ioana Maria Otilia Lică, Ana Maria Daviţoiu, Sorina Chindriş, Gabriela Vlad, Eugenia Buzoianu, Mirela Iancu, Ioana Adriana Ghiorghiu, Andrei Zamfirescu, Doina Anca Pleşca
Chiar şi în secolul XXI, bolile infectocontagioase sunt încă recunoscute pentru impactul asupra calităţii vieţii la populaţia p...
Utilizarea dispozitivelor electronice în prima copilărie (0-5 ani): beneficii educaționale, riscuri de neurodezvoltare și recomandări clinice bazate pe dovezi – o sinteză narativă
Adriana Mihai, Ileana-Katerina Ioniuc, Oana-Raluca Temneanu, Paula Popovici, Alice-Nicoleta Grudnicki, Alina Murgu
Copiii cu vârsta mai mică de 5 ani cresc imersați în medii digitale bazate pe ecrane într-o măsură fără precedent istoric....