Henoch-Schönlein purpura is the most common immune vasculitis in the pediatric population, with a maximum incidence in the 2-8 year age group. Skin lesions are pathognomonic and may be accompanied by articular, gastrointestinal, renal, or rarely neurological and testicular manifestations. The disease has a self-limiting character, the short-term prognosis being influenced by the severity of the digestive involvement (the degree of upper or lower digestive haemorrhage), and the long-term relationship is related to the degree of renal involvement.
STUDII CLINICE
Particularităţi clinico-biologice în purpura Henoch-Schönlein la copil
Clinical and biological features in Henoch-Schönlein purpura in children
Bogdan A. Stana,
Prof. dr. Evelina Moraru,
Alice Azoicăi,
Paula Popovici,
Irina Crișcov,
Ileana Ioniuc,
Monica Alexoae,
Mihai M. Hogaş,
Alina Murgu
First published: 27 noiembrie 2018
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Pedi.51.3.2018.2074
Abstract
Rezumat
Purpura Henoch-Schönlein este cea mai frecventă vasculită imună întâlnită în populaţia pediatrică, având o incidenţă maximă la grupa de vârstă 2-8 ani. Leziunile cutanate sunt patognomonice şi pot fi însoţite de manifestări articulare, gastrointestinale, renale sau, mai rar, neurologice şi testiculare. Boala are un caracter autolimitant, prognosticul pe termen scurt fiind influenţat de severitatea afectării digestive (gradul hemoragiilor digestive superioare sau inferioare), iar cel pe termen lung este în relaţie cu gradul afectării renale.
Purpura Henoch-Schönlein (PHS) este o tulburare mediată de imunoglobulina A (IgA), caracterizată printr-o vasculită generalizată care implică vasele mici ale pielii, ale tractului gastrointestinal, rinichilor, articulaţiilor şi, rareori, ale plămânilor şi ale sistemului nervos central (SNC). Este o afecţiune a vaselor mici (arteriole, venule, capilare), datorată depunerii în pereţii acestora a complexelor imune circulante, a imunoglobulinelor (Ig) A sau a fracţiunii 3 (C3) a complementului.
Purpura Henoch-Schönlein este cea mai frecventă vasculită imună întâlnită în populaţia pediatrică, având o incidenţă maximă în grupa de vârstă 2-8 ani. Leziunile cutanate sunt cele mai constante şi pot fi însoţite de manifestări articulare, gastrointestinale, renale sau, mai rar, neurologice şi testiculare. Boala are un caracter autolimitant, prognosticul pe termen scurt fiind influenţat de severitatea afectării digestive (gradul hemoragiilor digestive superioare sau inferioare), iar cel pe termen lung fiind în relaţie cu gradul afectării renale.
Epidemiologie
-
mai frecventă la copii decât la adulţi
-
vârsta: 4-7 ani, incidenţa maximă
-
predominanţă masculină
-
incidenţă sezonieră: crescută iarna şi primăvara
-
manifestări extracutanate:
-
articulare (75%)
-
digestive (50%)
-
-
afectare renală (20-90%) → IRC → dializă → transplant
-
mortalitate la copii: <1%, prin complicaţii digestive.
Etiologie
Etiologia exactă a purpurei Henoch-Schönlein nu este deocamdată cunoscută, însă se pare că există anumiţi factori de risc pentru apariţia acesteia, printre care se numără:
-
afecţiuni respiratorii superioare
-
vaccinări, inclusiv cele împotriva pojarului, febrei tifoide sau holerei
-
agresiunea unor agenţi infecţioşi, în special bacterii şi virusuri (streptococ, parvovirusuri)
-
contactul cu anumite insecte, alimente sau medicamente (antibiotice, antihistaminice).
Se crede că sistemul imunitar al organismului joacă un rol important în declanşarea mecanismelor patogenice implicate.
Un răspuns imun anormal la o infecţie poate fi un factor trigger în multe cazuri. Aproximativ două treimi dintre cazurile de PHS apar la câteva zile după apariţia simptomelor unei infecţii respiratorii superioare sau a altor afecţiuni infecţioase acute.
Ca posibili agenţi etiologici declanşatori ai purpurei Henoch-Schönlein se pot enumera:
Infecţii:
-
infecţiile tractului respirator superior
-
pojar
-
rubeolă
-
varicelă
-
parvovirus uman B19
-
adenovirusuri
-
virus Coxsackie
-
Mycoplasma pneumoniae
-
Salmonella hirschfeldii
-
Clostridium difficile
-
Morganella morganii
-
Streptococcus pneumoniae
-
tuberculoza
-
Legionella
-
Helicobacter pylori
-
Toxoplasma gondi
-
Entameoba hystolitica.
Medicamente:
-
vancomicina
-
ranitidina
-
streptokinaza
-
cefuroxima
-
diclofenacul
-
enalaprilul
-
captoprilul.
Anatomie patologică
Este o vasculită leucocitoclastică a vaselor mici, caracterizată la nivelul intestinului prin hemoragii submucoase, subseroase, iar la nivelul rinichilor, de glomerulonefrite focale sau difuze.
Caracteristic, se descrie existenţa depozitelor (complexe imune cu IgA) în vasele afectate, mezangiul glomerular şi piele. În vasele afectate se depun complexe imune (IgA, C3, properdina) şi polimorfonucleare (leucocitoclazie).
Patogeneza
Deşi patogeneza PHS rămâne, de asemenea, necunoscută, IgA joacă un rol critic în imunopatogeneza PHS, aşa cum reiese din creşterea concentraţiilor serice de IgA, a complexelor imune circulante care conţin IgA şi a depunerii de IgA în pereţii vaselor şi în celulele mesangiale extraglomerulare de la nivelul rinichilor.
Agregatele IgA sau complexele IgA cu complement depuse în organele-ţintă, care au ca rezultat elaborarea mediatorilor inflamatori, inclusiv a prostaglandinelor vasculare, cum este prostaciclina, pot juca un rol central în patogeneza vasculitei PHS.
Din punct de vedere histologic, PHS se caracterizează prin infiltrarea vaselor mici de sânge prin leucocite polimorfonucleare şi prin prezenţa leucocitoclasisului.
Imunofluorescenţa ţesuturilor poate dezvălui prezenţa depozitelor imune de IgA dominantă în pereţii vaselor mici (capilare, venule, arteriole sau glomeruli renali).
Diagnostic clinic
Erupţiile cutanate apar la 95-100% dintre pacienţi, cu localizare la nivelul membrelor inferioare şi al feselor în mod special. Pacientul se prezintă cu puncte de mici dimensiuni, palpabile, roşietice, care evoluează în timp spre o culoare purpurie, iar apoi vineţie. Purpura poate apărea şi la nivelul membrelor superioare, al cotului, al feţei şi al trunchiului şi e mai gravă în zonele de presiune, de exemplu în zona taliei.
Edem subcutanat (20-50%).
Durerile articulare apar (60-80%) mai ales la nivelul genunchilor, gleznei şi coatelor, dar şi la încheieturile mâinilor şi picioarelor. Inflamaţia articulară, care implică durere şi umflături, apare în aproximativ trei sferturi dintre cazuri, afectând în special genunchii şi gleznele. De obicei durează doar câteva zile şi nu cronicizează. Artralgia şi/sau artrita sunt însoţite de tumefierea şi sensibilitatea ţesutului moale din apropierea şi din jurul articulaţiilor afectate. La copiii foarte mici, tumefierea ţesutului moale de la nivelul mâinilor, picioarelor, al frunţii şi al scrotului poate apărea precoce în cursul bolii.
Durerile abdominale apar la mai mult de jumătate dintre persoanele cu PHS, sunt intermitente, sunt resimţite în jurul ombilicului şi pot fi însoţite de hemoragie gastrointestinală uşoară sau severă. Inflamaţia tractului gastrointestinal poate provoca dureri sau crampe; aceasta poate duce, de asemenea, la pierderea apetitului, vărsături, diaree şi, ocazional, sânge în scaun. În unele cazuri, pacienţii pot avea dureri abdominale înainte de apariţia erupţiilor cutanate. În cazuri rare poate apărea invaginaţia intestinală, urgenţă chirurgicală.
Afectarea renală asociată PHS (peste 40%) se manifestă prin hematurie, modificându-se culoarea urinei într-un roşu-cărămiziu. La majoritatea pacienţilor, insuficienţa renală este uşoară şi dispare fără consecinţe pe termen lung. Este important să se monitorizeze îndeaproape funcţia renală, deoarece aproximativ 5% dintre pacienţi pot dezvolta o boală renală progresivă, iar 1% dintre pacienţi evoluează către insuficienţă renală cronică. Uneori poate apărea chiar şi sindromul nefrotic, care se manifestă prin edeme generalizate ca urmare a hipoproteinemiei.
Criterii de diagnostic
1. Purpura palpabilă: prezenţa leziunilor cutanate hemoragice nelegate de trombocitopenie
2. Vârsta < 20 ani la debutul bolii
3. Angor intestinal: dureri abdominale difuze, agravate după mese, sau ischemie intestinală, de obicei cu diaree sangvinolentă
4. Infiltrat granulocitar în pereţii vasculari: biopsie cu PMN în pereţii arteriolelor sau venulelor.
Explorări paraclinice în PHS
Diagnosticul de PHS este mai ales clinic şi se bazează pe prezenţa erupţiei purpurice clasice, limitată de obicei la membrele inferioare şi la fese.
Pe lângă leziunea tegumentară specifică, purpura se asociază cel puţin cu una dintre manifestările următoare: durere abdominală, afectare articulară (artrită sau artralgie) şi afectare renală (cel mai des hematuria).
Pentru un diagnostic corect se recomandă o biopsie tegumentară, unde pot fi găsite depozite de IgA. Dacă semnele şi simptomele sunt evidente, biopsia nu este necesară, mai ales la copii. În afectarea renală, biopsia renală poate fi necesară pentru evidenţierea gradului alterării şi pentru prognosticul bolii. Testele sangvine de rutină pun în evidenţă semnele de infecţie, anemie sau afectarea renală. Se pot efectua examene imagistice, ca ecografia şi radiografia abdominală, dacă durerea abdominală este severă.
Pentru PHS nu există teste de laborator specifice, însă pot apărea următoarele modificări:
-
trombocitoză moderată
-
leucocitoză moderată
-
VSH crescut
-
anemie datorată sângerării intestinale acute sau cronice
-
CIC prezente
-
IgA şi IgM crescute.
În caz de invaginaţie intestinală, clisma baritată are atât rol diagnostic, cât şi terapeutic.
Diagnostic diferenţial
Vasculita Wegener şi poliangeita microscopică sunt principalele afecţiuni care au semne şi simptome asemănătoare cu PHS. Acestea prezintă de asemenea purpură, dureri articulare şi afectare renală.
Alte diagnostice diferenţiale ale PHS includ:
-
abdomenul acut chirurgical
-
infarctul intestinal sau perforaţia intestinală
-
reacţiile adverse la medicamente
-
alte patologii renale cronice
-
vasculita leucocitoclastică
-
glomerulonefrita acută
-
insuficienţa renală acută
-
endocardita bacteriană
-
abuzul şi neglijarea copilului (abuz fizic)
-
coagularea intravasculară diseminată
-
purpura trombocitopenică idiopatică
-
boala inflamatorie cronică a intestinului
-
meningita
-
infecţii meningococice
-
pancreatita acută
-
varicela
-
gastroenterita acută
-
sângerarea gastrointestinală
-
invaginaţia intestinală
-
boala Kawasaki
-
encefalita
-
febra reumatică
-
artrita reumatoidă
-
şocul septic
-
lupus eritematos sistemic
-
torsiunea testiculară.
Managementul PHS
PHS este o boală autolimitantă şi nu necesită un tratament specific pentru a scurta durata simptomelor. Controlul simptomatic al bolii include hidratarea adecvată, repausul la pat şi măsuri farmacologice nespecifice, dacă este cazul, în caz de apariţie a complicaţiilor bolii sau a determinărilor multiple (articulare, digestive, renale).
Tratamentul formei fără afectare renală constă în repaus la pat în faza acută, antibiotice numai în cazul în care s-a dovedit infecţia de focar; antiinflamatoare nesteroidiene (aspirină/ibuprofen etc.) sau antalgice. În cazul durerilor abdominale colicative se poate administra prednison în cură scurtă (2-3 săptamâni) sau metilprednisolon (3 pulsaţii i.v.), asociat cu inhibitori de pompă de protoni (omeprazol) sau inhibitori H2 (ranitidină/famotidină etc.) Corticoterapia nu previne dezvoltarea nefropatiei.
Afectarea renală sub formă de hematurie microscopică fără afectarea funcţiei renale nu impune tratament agresiv cu imunosupresoare sau corticoterapie. În aceste situaţii, bolnavii vor beneficia individualizat de antiagregante plachetare sau agenţi hipotensivi (dacă se impune).
Nefropatia demonstrată numai prin PBR impune un tratament agresiv şi precoce cu imunosupresive frecvent în asociere: pulsaţii (i.v.) cu metilprednisolon (mai ales în glomerulonefrita membrano-proliferativă crescentică), asociate cu ciclofosfamidă, sau azathioprină, sau ciclosporină A, sau mycophenolat mofetil. În formele severe, pot fi utile doze mari de imunoglobulină (i.v.).
Transplantul renal este măsura ultimă de rezervă, în caz de insuficienţă renală cronică neresponsivă la terapia medicamentoasă, unele studii menţionând totuşi riscul recurenţei nefritei posttransplant.
Tratamentul chirurgical rămâne de elecţie în cazurile cu complicaţii digestive severe (perforaţie, invaginaţie etc.).
Complicaţii
În cazul majorităţii pacienţilor, simptomele purpurei Henoch-Schönlein se ameliorează de la sine în câteva săptămâni şi dispar fără să lase în urmă alte complicaţii. Recurenţele sunt însă destul de frecvente. Cei mai predispuşi sunt copiii care au avut încă de la început o stare mai gravă şi purpura extinsă. Chiar şi în condiţiile apariţiei recurenţelor, acestea au o intensitate moderată comparativ cu episodul iniţial.
În cazuri rare, purpura se poate complica cu obstrucţie intestinală care reduce fluxul sangvin local şi se poate complica cu fenomene ischemice intestinale şi chiar pancreatice.
Cea mai importantă complicaţie a purpurei este reprezentată de afectarea renală, care se manifestă prin hematurie, proteinurie, edeme generalizate şi hipertensiune arterială. În cazurile grave, afecţiunea renală acută poate să evolueze spre boala renală cronică, iar pacientul poate avea indicaţie de transplant renal sau dializă.
Prognostic
Purpura Henoch-Schönlein este o afecţiune în general benignă şi are un prognostic bun. Peste 80% dintre pacienţi se confruntă doar cu un episod izolat, care durează câteva săptămâni, restul având şi recurenţe (mai frecvent apar la copii).
Studiu – particularităţi clinico-biologice ale purpurei Henoch-Schönlein la copiii internaţi în Clinica II Pediatrie, Iaşi
Lotul de studiu a fost constituit din 12 copii diagnosticaţi cu purpură Henoch-Schönlein, internaţi şi monitorizaţi în Clinica II Pediatrie – Spitalul Clinic de Urgenţă pentru Copii „Sf. Maria”, Iaşi, în perioada ianuarie–august 2018.
Criteriul de selecţie şi includere a pacienţilor în studiu l-a constituit existenţa diagnosticului de purpură Henoch-Schönlein între diagnosticele de externare.
Modelul studiului efectuat a fost retrospectiv. Componenta retrospectivă a fost realizată prin consultarea foilor de observaţie care au oferit date anamnestice, clinice şi de laborator. Pacienţii nou diagnosticaţi au fost monitorizaţi prospectiv, prin măsurarea a diverşi parametri clinici şi biologici după instituirea tratamentului igieno-dietetic şi patogenic şi consilierea de specialitate.
Metodologia de lucru a cuprins date referitoare la anamneză, existenţa factorilor de risc, momentul diagnosticului, evoluţia bolii, parametri clinici şi biologici semnificativi, tratamentul urmat şi date despre calitatea vieţii.
Rezultate şi discuţii
Vârsta medie a pacienţilor la momentul diagnosticului a fost de 5,3 ani, cu valori cuprinse între 1 şi 9 ani la internarea în clinică. După cum se ştie, PHS este apanajul primei copilării şi al vârstei preşcolare, dar se observă că au existat şi cazuri cu vârste mai mari (trei cazuri cu vârste între 8 şi 9 ani).
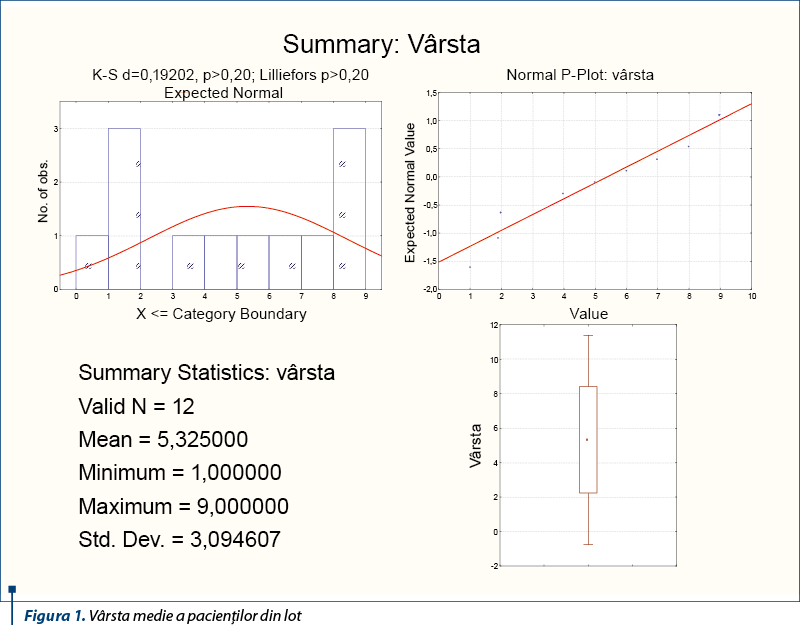
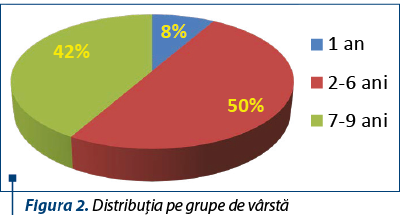
Distribuţia pacienţilor în funcţie de vârsta la diagnostic este redată în figura 2. Se observă că jumătate dintre pacienţii din lotul de studiu au vârstele cuprinse între 2 şi 6 ani, corespunzător vârfului maxim de incidenţă a bolii la vârsta pediatrică.
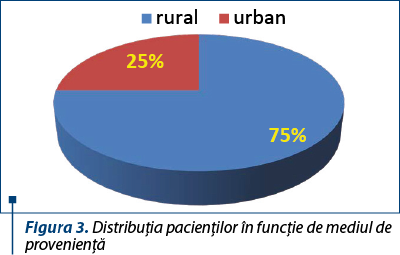
În ceea ce priveşte provenienţa pacienţilor, majoritatea au provenit din mediul rural (trei sferturi dintre pacienţi).
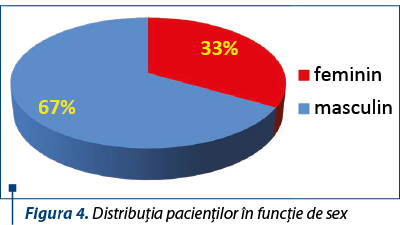
În funcţie de distribuţia pacienţilor pe sexe, se observă că predomină băieţii, ceea ce concordă cu datele din literatură – raport de 2:1 masculin:feminin.
În ceea ce priveşte comorbidităţile pacienţilor, se observă că artropatia reactivă predomină, deoarece reprezintă o inflamaţie a articulaţiilor, iar în PHS articulaţiile mari sunt cel mai frecvent implicate, singurele simptome fiind durerea şi edemul. S-a dovedit şi o frecvenţă crescută a celor care asociază sindrom dureros abdominal, faringoamigdalită acută, deficit de IgG şi anemie.

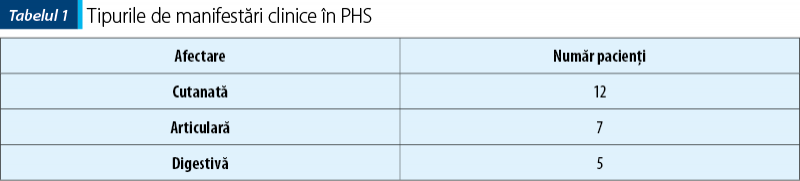
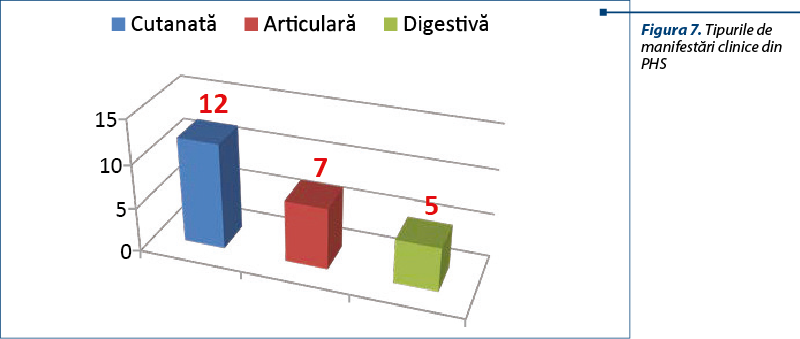
Din punctul de vedere al manifestărilor clinice, erupţia purpurică este cea care reprezintă de cele mai multe ori motivul de prezentare la medic. Trebuie diferenţiat între purpura din PHS, purpura trombocitopenică (când valorile trombocitelor sunt scăzute), purpura din sindroamele toxiinfecţioase (copil cu stare generală foarte gravă, febră înaltă, sindrom toxico-septic clinic şi biologic) sau purpura din alte afecţiuni.
Un element deosebit de important îl constituie anamneza, când medicul regăseşte un episod infecţios în antecedentele recente (infecţie respiratorie superioară sau inferioară, infecţie digestivă, urinară etc.). De aceea, anamneza atentă este deosebit de importantă.
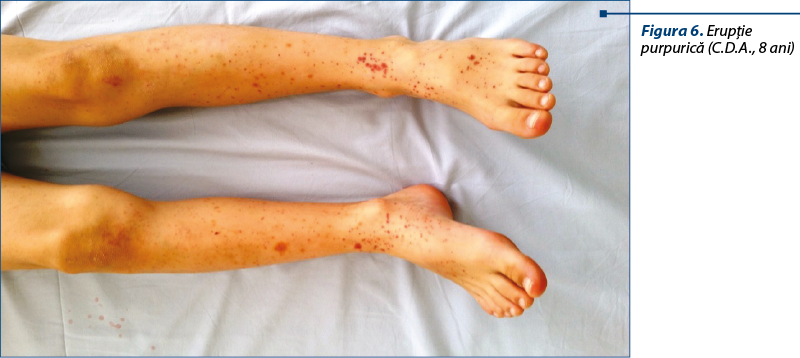
După cum se poate observa, toţi copiii din lotul studiat au prezentat erupţie purpurică metamerică, simetrică, distribuită decliv la membrele inferioare şi uneori pe fese. Examinarea pacientului trebuie făcută în lumină corespunzătoare (lumina zilei), cu examenul atent al tegumentelor, pentru a observa distribuţia şi caracterul peteşiilor.
În PHS, erupţia purpurică poate apărea în valuri, de aceea pacientul trebuie examinat în fiecare zi, dezbrăcat complet, pentru a surprinde eventualele elemente purpurice noi, care sunt semn important de recădere a bolii.
Din lotul studiat, 7 copii au prezentat în evoluţie recăderi, adică un procent de 58,3% dintre cazuri au prezentat recăderi ale bolii.
Afectarea articulară se observă prin tumefacţii articulare, tulburări de mers şi semne inflamatorii locale articulare (roşeaţă, căldură, tumefiere, durere articulară). Aceste elemente nu se observă direct uneori, de aceea asistenta medicală trebuie să le cunoască şi să le caute. În momentul în care există aceste elemente clinice, vor fi aduse la cunoştinţa medicului curant.
Din cazurile descrise, la un număr de 7 copii (58,3%) s-au descris afectări articulare, manifestate prin semne inflamatorii locale şi impotenţă funcţională. La cinci dintre aceştia, afectarea articulară a apărut după purpură.
Se observă faptul că toţi pacienţii prezintă manifestări cutanate, iar mai mult de jumătate sunt afectaţi şi articular.
Afectarea digestivă este nespecifică, din cauza vasculitei de la nivelul capilarelor intestinale. În acest caz, copilul va prezenta dureri abdominale nespecifice, inapetenţă, uneori greaţă şi vărsături. Când copilul varsă, asistenta medicală trebuie să aprecieze caracteristicile vărsăturii, iar prezenţa de striuri de sânge reprezintă un element de gravitate maximă şi se va aduce imediat la cunoştinţa medicului de gardă sau a medicului curant.
Un element deosebit de important îl reprezintă aspectul scaunelor. Asistenta medicală va urmări scaunele ca frecvenţă, consistenţă şi aspect. Existenţa striurilor de sânge sau a scaunelor melenice reprezintă un element de gravitate. Totodată, aspectul nedigerat al scaunelor poate reprezenta consecinţa vasculitei intestinale, cauzată de edem. Tot ca manifestare digestivă, se citează şi inapetenţa, însoţită de dureri abdominale.
Un copil din lotul studiat a fost internat iniţial pentru dureri abdominale nespecifice, suspicionându-se debutul unei gastroenterite, dar fără scaune modificate, copilul fiind inapetent şi ulterior dezvoltând deshidratare acută prin lipsă de aport. În 24 de ore durerile abdominale se accentuează, dezvoltă abdomen acut chirurgical şi necesită laparoscopie exploratorie care evidenţiază invaginaţie intestinală. Intraoperator se descriu elemente purpurice şi la nivelul anselor intestinale invaginate. După cura chirurgicală a invaginaţiei, la revenirea în Clinica II Pediatrie se evidenţiază şi elementele purpurice cutanate. Evoluţia clinică şi biologică a fost favorabilă pe termen scurt şi lung, dar invaginaţia intestinală cu sindromul de abdomen acut chirurgical a fost particularitatea acestui caz de PHS.
Un alt copil din lotul studiat prezenta iniţial o formă de PHS cu evoluţie autolimitată, primul episod cedând relativ repede la corticoterapie, calciterapie şi repaus la pat. La 10 zile de la externare, copilul se prezintă în UPU Chirurgie pentru tumefacţie dureroasă de hemiscrot stâng, diagnosticul fiind de orhiepididimită acută după PHS, care a necesitat cura chirurgicală şi urmărirea chirurgicală a acestui caz.
Prin urmare, tabloul clinic al PHS este deosebit de variabil, iar asistenta medicală trebuie să urmărească îndeaproape pacientul, orice nou simptom sau semn apărut trebuind interpretat rapid în context clinic.
Tabloul biologic
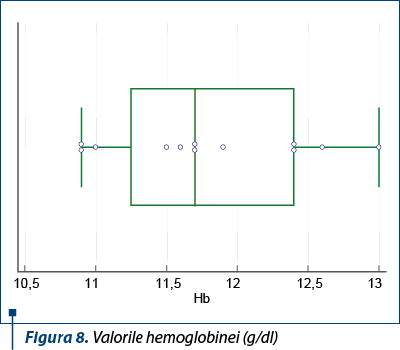
Inflamaţia a fost prezentă la toţi copiii din lot, atât VSH, cât şi CRP şi fibrinogenul având valori crescute.
Anemia a fost regăsită la 7 pacienţi, în grade uşoare, doi dintre aceştia prezentând şi hemoragie digestivă inferioară (scaune cu sânge, remise la tratament conservator). Într-un caz cu PHS şi dureri abdominale, ecografia abdominală şi examenul chirurgical au confirmat invaginaţia jejuno-ileală, iar prin laparotomie s-a rezolvat invaginaţia şi s-a constatat şi ileita terminală.
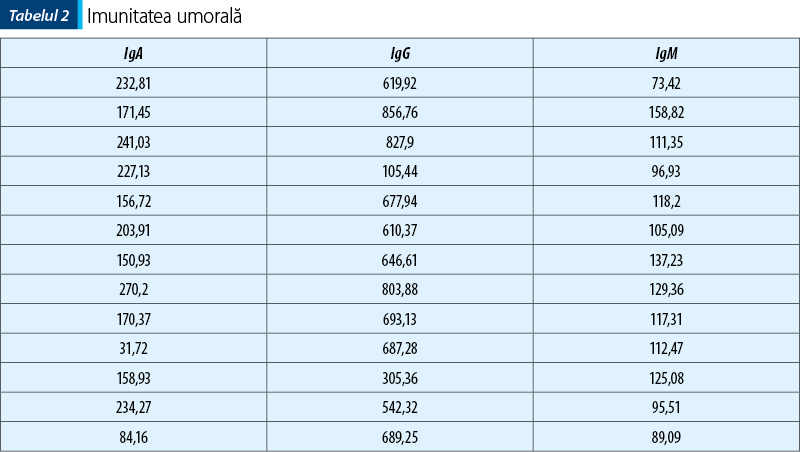
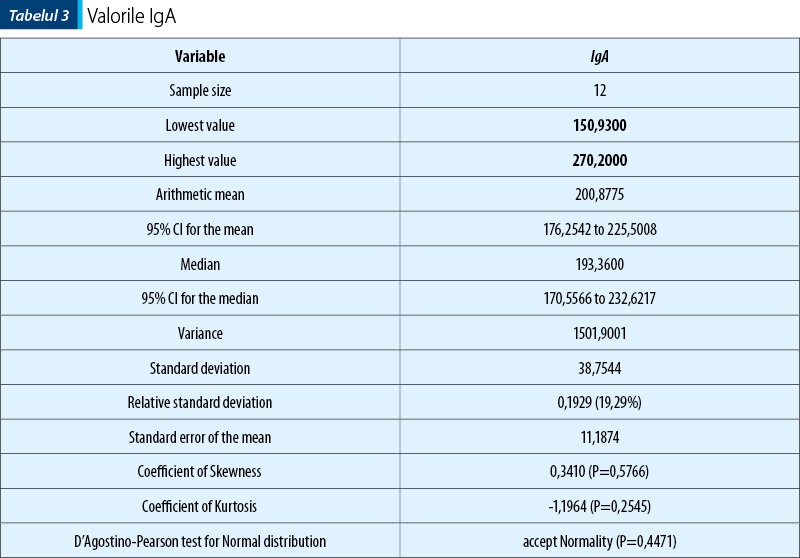
În ceea ce priveşte nivelul IgA, acesta a fost regăsit crescut la jumătate din pacienţii din lot (tabelul 2).
Tratament
Ca tratament igienico-dietetic, se recomandă copilului:
-
Repaus la pat – se recomandă repaus la pat pacientului cu PHS deoarece, punând presiune pe membrele inferioare, capilarele din această zonă se sparg şi astfel apar peteşii; clinostatismul îmbunătăţeşte circulaţia sângelui la nivel renal şi poate grăbi vindecarea leziunilor renale, însă dacă pacientul menţine timp îndelungat aceeaşi poziţie, este predispus la apariţia unor complicaţii (escare), care îi împiedică procesul de vindecare. Pentru prevenirea apariţiilor acestora, se recomandă schimbarea poziţiei bolnavului.
-
Aport alimentar cantitativ şi calitativ optim.
-
Se vor evita alimentele alergenice, aditivii alimentari, conservanţii de orice fel.
-
Regim hiposodat asociat corticoterapiei.
Tratamentul formei fără afectare renală constă în: repaus la pat în faza acută, antibiotice numai în cazul în care s-a dovedit infecţia de focar; antiinflamatoare nesteroidiene (aspirină/ibuprofen etc.) sau antalgice; în cazul durerilor abdominale colicative se poate administra prednison 2 mg/kg/zi po, în cură scurtă (2-3 săptămâni) sau metilprednisolon (3 pulsaţii i.v.) asociat cu inhibitori de pompă de protoni (omeprazol) sau inhibitori H2 (ranitidină/famotidină etc.). Corticoterapia nu previne dezvoltarea nefropatiei.
-
Afectarea renală sub formă de hematurie microscopică, fără afectarea funcţiei renale, nu impune tratament agresiv cu imunosupresoare sau corticoterapie. În aceste situaţii, bolnavii vor beneficia individualizat de: antiagregante plachetare (dipiridamol – 5 mg/kg/zi); agenţi hipotensivi (dacă se impune, în faza acută de glomerulonefrită).
-
Nefropatia, demonstrată numai prin puncţie-biopsie renală, impune un tratament agresiv şi precoce cu imunosupresive frecvent în asociere: pulsaţii (i.v.) cu metilprednisolon (mai ales în glomerulonefrita membrano-proliferativă crescentică) asociate cu ciclofosfamidă, sau azathioprină, sau ciclosporină A, sau micofenolat mofetil. În formele severe, pot fi utile doze mari de imunoglobulină (i.v.).
-
Transplantul renal este măsura ultimă de rezervă, în caz de insuficienţă renală cronică (IRC) neresponsivă la terapia medicamentoasă, unele studii menţionând totuşi riscul recurenţei nefritei posttransplant.
Evoluţie şi prognostic
Evoluţia naturală a bolii este autolimitată, aproximativ un an, cu recăderi şi remisiuni, dar cu vindecare deplină ulterioară, fără sechele în 95-96% dintre cazuri. Recăderile apar în circa 50% din cazuri, cu o medie de două-trei puseuri după debut care succed la un interval aproximativ de 3-4 săptămâni, cu o evoluţie spre ameliorare în 6-8 săptămâni. La copilul mic, intensitatea şi durata acestora este mai scăzută faţă de debut şi faţă de copilul mare. Rar apar recăderi la un an după debut, spontan sau favorizate cel mai frecvent de infecţii respiratorii.
Morbiditatea prin boală este determinată de afectarea renală severă (IRC), în 1-3% din cazuri. Mortalitatea, reprezentând 0,5-2% din cazuri, în faza acută a bolii, este cauzată de complicaţii digestive severe (obstrucţie intestinală) sau renale (nefrită acută +/- HTA/convulsii), iar la distanţă, decesul este prin insuficienţă renală cronică terminală.
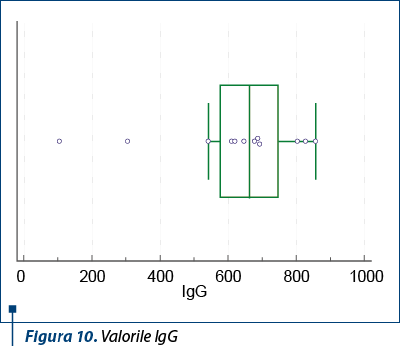
Complicaţia renală determină prognosticul bolii. Afectarea renală prezentă de la debut sau în primele trei luni de evoluţie, ca şi prezenţa constantă a exacerbărilor însoţite de simptome renale reprezintă un indicator de prognostic nefavorabil pentru persistenţa afectării renale ulterioare.
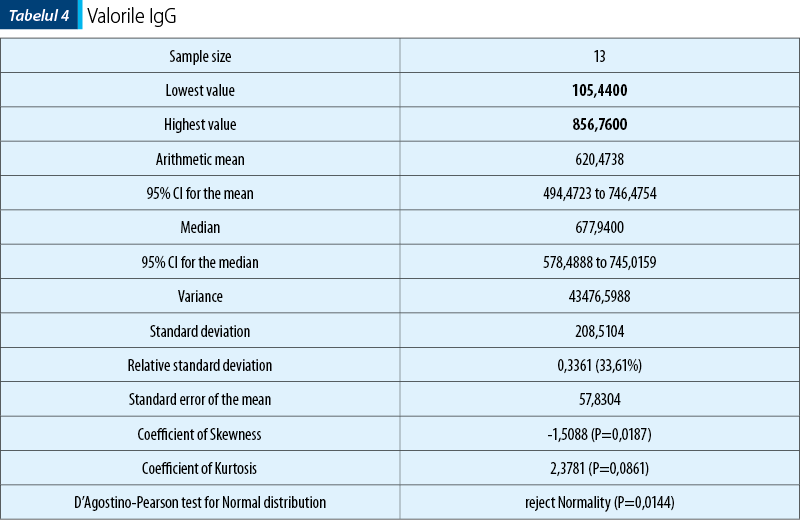
Evoluţiile clinice ale pacienţilor din lot au fost în general favorabile, din cei 12 copii doar doi prezentând recăderi ale PHS, la 4 luni, respectiv 6 luni. Afectarea renală nu a fost regăsită la niciun pacient dintre cei 12 pe parcursul perioadei de studiu (proba Addis şi parametrii renali nemodificaţi). Aceşti pacienţi sunt încă în urmărire pe o perioadă de cel puţin un an, pentru a surprinde complicaţia renală, care stabileşte prognosticul bolii.
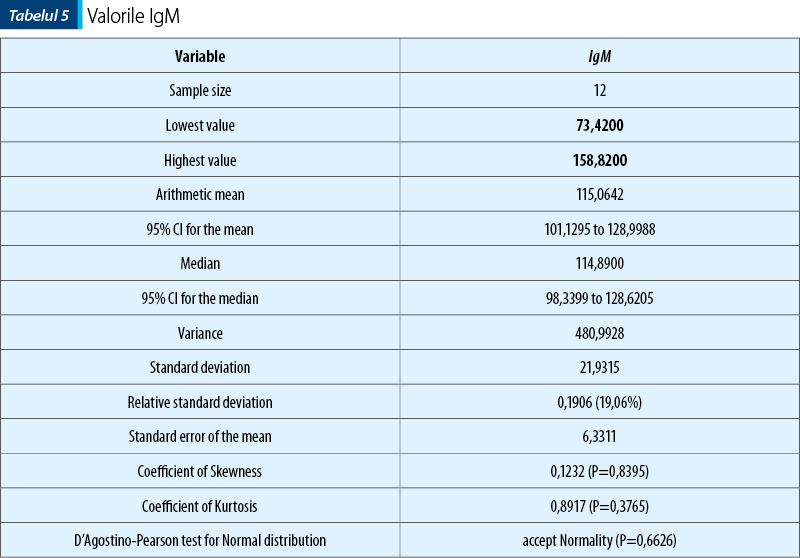
Concluzii
Purpura Henoch-Schönlein reprezintă o vasculită imună frecventă în practica pediatrică, care necesită recunoaştere rapidă şi diagnostic diferenţial atent. Afectarea cutanată este primordială, urmată de afectarea articulară şi digestivă. Complicaţiile acute ale bolii sunt multiple, iar semnele precoce ale complicaţiilor trebuie surprinse cât mai rapid. Pe termen lung, afectarea renală este cea care determină prognosticul bolii.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
- Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, García-Fuentes M, González-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997 May; 40(5):859-64.
- Szer IS. Henoch-Schönlein purpura. Curr Opin Rheumatol. 1994 Jan; 6(1):25-31.
- Chen O, Zhu XB, Ren P, Wang YB, Sun RP, Wei DE. Henoch Schonlein Purpura in children: clinical analysis of 120 cases. Afr Health Sci. 2013 Mar; 13(1):94-9.
- Bowman P, Quinn M. Question 1: Should steroids be used to treat abdominal pain caused by Henoch-Schonlein purpura?. Arch Dis Child. 2012 Nov; 97(11):999-1000.
- Calvo-Rio V, Hernandez JL, Ortiz-Sanjuan F, et al. Relapses in patients with Henoch-Schonlein purpura: Analysis of 417 patients from a single center. Medicine (Baltimore). 2016 Jul; 95 (28):e4217.
- Narchi H. Risk of long term renal impairment and duration of follow up recommended for Henoch-Schonlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005 Sep; 90(9):916-20.
- McCarthy HJ, Tizard EJ. Clinical practice: Diagnosis and management of Henoch-Schönlein purpura. Eur J Pediatr. 2010 Jun; 169(6):643-50.
Articole din ediţiile anterioare
STUDII CLINICE | Ediţia 2 46 / 2017
Purpură Henoch-Schönlein. Prezentare de caz
Laurențiu B. Dimache, Alexandra Sîrghi, Prof. dr. Evelina Moraru
Purpura Henoch-Schönlein este cea mai frecventă vasculită imună întâlnită în populația pediatrică, având o incidență maximă în grupa de vârstă ...
07 iunie 2017
CASE REPORTS | Ediţia 2 58 / 2020
Aspect particular al purpurei Henoch-Schönlein la copil – prezentare de caz
Alice Azoicăi, Maria-Ruxandra Cepoi, Bogdan A. Stana
Purpura Henoch-Schönlein este o vasculită leucocitoclazică ce este caracterizată printr-o angeită a vaselor mici indusă prin mecanism imunolog...
23 mai 2020