Imunodeficienţa comună variabilă – fenotipuri clinice şi metode de evaluare paraclinică
Common variable immunodeficiency – clinical phenotypes and paraclinical workup
Abstract
Primary immunodeficiencies, or inborn errors of immunity, comprise a heterogeneous group of conditions characterized by the deficient or absent functioning of one or more components of the immune system. They predispose to frequent and severe infections, autoimmunity, alteration of immune homeostasis, autoinflammatory diseases, lymphoproliferative diseases, neoplasias and, according to the latest classification published in 2022, bone marrow failure. Although considered rare diseases, they have a higher prevalence than initially thought.This paper reviews predominantly antibody immunodeficiencies and addresses common variable immunodeficiency (CVID) as a central theme from the perspective of clinical phenotypes, diagnostic investigation methods, and post-diagnostic monitoring of the CVID patient.
Keywords
primary immunodeficiencyinborn errors of immunitycommon variable immunodeficiencyclinical phenotypesRezumat
Imunodeficienţele primare, sau erorile înnăscute ale imunităţii, cuprind un grup eterogen de afecţiuni caracterizate prin funcţionarea deficitară sau absentă a uneia sau a mai multor componente ale sistemului imunitar. Acestea predispun la infecţii frecvente şi severe, autoimunitate, alterarea homeostaziei imune, boli autoinflamatorii, boli limfoproliferative, neoplazii şi, conform ultimei clasificări publicate în 2022, insuficienţă medulară. Deşi considerate boli rare, acestea au o prevalenţă mai mare decât cea considerată iniţial. Această lucrare reprezintă o trecere în revistă a imunodeficienţelor predominant anticorpice şi abordează ca temă centrală imunodeficienţa comună variabilă (CVID), din perspectiva fenotipurilor clinice şi a metodelor de investigare cu scop diagnostic şi de monitorizare postdiagnostică a pacientului cu CVID.Cuvinte Cheie
imunodeficienţă primarăerori înnăscute ale imunităţiiimunodeficienţă comună variabilăfenotip clinicScopul lucrării
Acest articol reprezintă o trecere în revistă a imunodeficienţelor predominant anticorpice şi abordează ca temă centrală imunodeficienţa comună variabilă (CVID), din perspectiva fenotipurilor clinice şi a metodelor de investigare cu scop diagnostic şi de monitorizare postdiagnostică a pacientului cu CVID.
Imunodeficienţele primare – definiţie, clasificare, date epidemiologice
Imunodeficienţele primare (PIB), sau erorile înnăscute ale imunităţii, cuprind un grup eterogen de afecţiuni caracterizate prin funcţionarea deficitară sau absentă a uneia sau a mai multor componente ale sistemului imunitar. Acestea predispun la infecţii frecvente şi severe, autoimunitate, alterarea homeostaziei imune, boli autoinflamatorii, boli limfoproliferative, neoplazii şi, conform ultimei clasificări publicate în 2022, insuficienţă medulară(1).
Începând cu 1990, Uniunea Internaţională a Societăţilor de Imunologie (IUIS; International Union of Immunological Societies) şi Comitetul Experţilor în Imunodeficienţe Primare (EC; PID Expert Committee), denumite acum Comitetul Mutaţiilor Înnăscute de Imunitate (Inborn Errors of Immunity Committee), publică, la fiecare doi ani, clasificarea revizuită a erorilor de imunitate moştenite. Societatea Europeană pentru Imunodeficienţe (ESID; European Society for Immunodeficiencis) colectează date din peste 126 de centre europene şi raportează peste 28000 de cazuri de imunodeficienţă primară(1).
Anul 2022 aduce ultima clasificare a erorilor înnăscute ale imunităţii/imunodeficienţelor primare, elaborată de International Union of Immunological Societies Expert Committee. Conform acesteia, grupul imunodeficienţelor primare umane cuprinde 485 de afecţiuni distincte, ca urmare a descoperirii unui număr de 55 de mutaţii noi faţă de clasificarea precedentă, publicată în 2020(1,3).
Conform ultimei clasificări a PID, acestea pot fi:
- imunodeficienţe combinate
- imunodeficienţe combinate cu trăsături de sindrom
- imunodeficienţe predominant anticorpice
- afecţiuni cu anomalii ale reglării imune
- defecte congenitale ale fagocitelor
- defecte prin funcţionarea intrinsecă a imunităţii înnăscute
- boli autoinflamatorii
- deficite ale sistemului complement
- insuficienţă medulară
- fenocopii ale erorilor înnăscute ale imunităţii.
Nu de puţine ori, acestea sunt intricate şi se suprapun la acelaşi individ(1). Detalierea acestora nu face obiectul lucrării de faţă; pentru o mai bună înţelegere a fiecăreia dintre acestea, autorii recomandă studiul ultimului ghid de imunodeficienţă primară(1).
Iniţial, imunodeficienţele primare au fost considerate boli rare, prevalenţa raportată fiind de 1/10000-1/50000 de naşteri. Ca urmare a progreselor privind caracterizarea fenotipurilor clinice şi a elucidării defectelor genetice implicate, în prezent se estimează că prevalenţa globală a imunodeficienţelor primare este de cel puţin 1/1000-1/5000 de naşteri. Deficitul imun selectiv de IgA este cea mai frecventă imunodeficienţă primară, urmat de imunodeficienţa comună variabilă (CVID; Common Variable Immunodeficiency) şi de agamaglobulinemia Bruton. Se apreciază că, la nivel mondial, aproximativ şase milioane de persoane suferă de o formă de imunodeficienţă primară(2,4).
Imunodeficienţe predominant anticorpice (umorale)
Tabelul 1 (A-D) redă, aşa cum se regăseşte în ,,Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee”, imunodeficienţele predominant anticorpice(1).
În plus faţă de mutaţiile monogenice care stau la baza acestor afecţiuni, unii indivizi cu XLP (X linked proliferative disease), sindrom WHIM (veruci, hipogamaglobulinemie, infecţii, mielocatexis), ICF (imunodeficienţă cu instabilitatea centromerilor şi anomalii faciale), VODI (imunodeficienţă cu boală hepatică veno-ocluzivă), sindrom Good (timom cu imunodeficienţă) sau cu mielodisplazie se adresează adesea de la început imunologului pentru infecţii recurente, hipogamaglobulinemie şi număr redus/normal de limfocite B. CVID include o serie de fenotipuri clinice şi de laborator distincte, care pot fi cauzate de diferiţi factori genetici şi/sau de mediu. O parte dintre indivizii cu CVID pot prezenta un număr foarte scăzut al limfocitelor B şi hipogamaglobulinemie şi niciun defect genetic care să fie pus în evidenţă cu resursele disponibile în prezent. Identificarea unei mutaţii la pacientul cu CVID poate fi utilă în alegerea tratamentului ţintit(1).
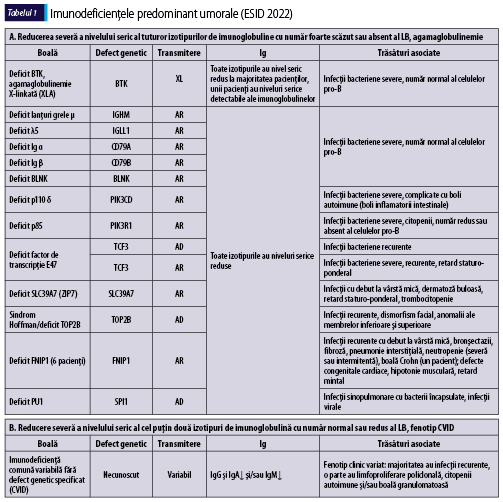
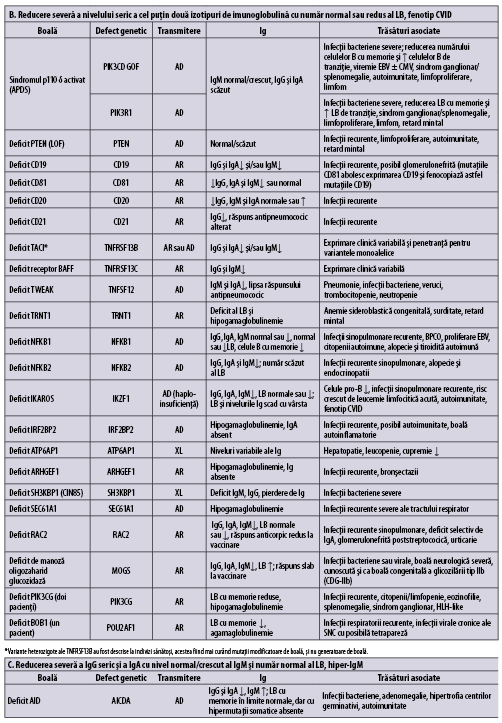
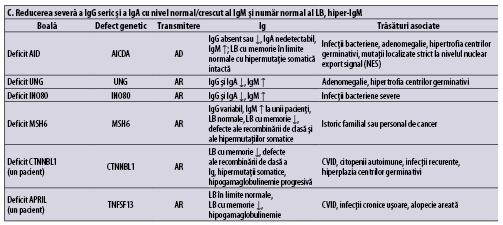
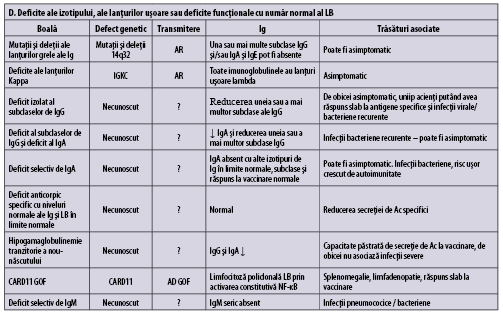
Imunodeficienţa comună variabilă
Date generale
CVID este o formă severă de imunodeficienţă primară predominant umorală, cu expresie variabilă (afectarea predominant a funcţiei şi/sau numărului limfocitelor B şi asociere, în unele cazuri, cu defecte ale limfocitelor T), cu fenotipuri clinice şi etiologii diferite. Cel mai adesea, CVID se exprimă clinic prin fenotipul infecţiilor repetate, însă la o parte din pacienţi tabloul clinic este dominat de complicaţii neinfecţioase, precum autoimunitate, afectare digestivă, boală pulmonară cronică, boală granulomatoasă, hiperplazie limfoidă, splenomegalie sau neoplazii(5). Riscul de mortalitate şi de morbiditate este mai crescut în categoria pacienţilor cu fenotip CVID cu complicaţii neinfecţioase(5).
CVID este imunodeficienţa primară umorală simptomatică a adultului cu cea mai mare prevalenţă, cu debut adesea la vârsta de adult (vârf al prevalenţei în decadele 3-4), cu o prevalenţă estimată la 1/25000 de indivizi. Fenotipurile clinice sunt variate, reunite de criteriul hipogamaglobulinemiei. O serie de anomalii ale răspunsului imun sunt asociate hipogamaglobulinemiei şi variază de la autoimunitate la neoplazii(5).
Diagnostic
Conform celui mai recent ghid ICON (International Consensus Document), diagnosticul de CVID respectă următoarele cinci coordonate(6):
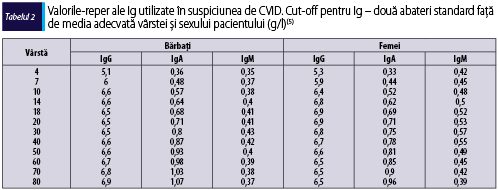
- Scăderea nivelului seric al IgG. Valorile standard ale imunoglobulinelor utilizate în evaluarea pacientului cu suspiciune de CVID sunt redate în tabelul 2. Este necesară obţinerea valorilor reduse semnificativ ale IgG (raportat la vârsta şi sexul pacientului – conform tabelului 2) cu ocazia a două măsurători la distanţă de cel puţin trei săptămâni; excepţie fac pacienţii cu niveluri foarte scăzute (<100-300 mg/dl, în funcţie de vârstă).
- Nivel seric scăzut al IgM sau al IgA.
- Răspuns alterat la vaccinare.
- Vârsta de peste 4 ani.
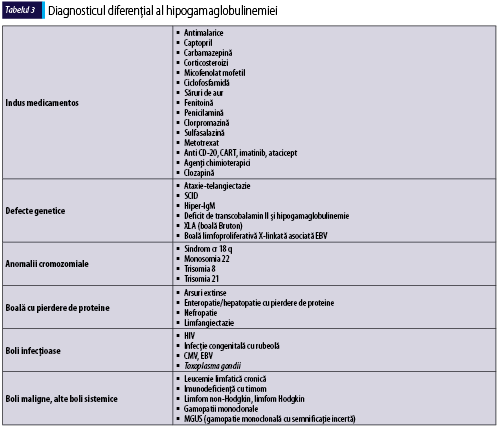
- Au fost excluse cauzele unei hipogamaglobulinemii secundare.
Criteriile de diagnostic al ESID aduc o serie de diferenţe faţă de ghidul ICON, respectiv(7):
- Scăderea IgA este criteriu obligatoriu.
- Un nivel redus al celulelor B cu memorie switched (<70% din valoarea prezisă pentru vârsta pacientului). Acesta poate să înlocuiască măsurarea titrului de anticorpi la vaccinare.
- Fără deficit marcant al limfocitelor T.
- Manifestări clinice de boală, precum susceptibilitate crescută pentru infecţii, semne şi simptome de boală autoimună, boală granulomatoasă sau limfoproliferare policlonală ori un membru al familiei cunoscut cu imunodeficienţă umorală (figura 1).
Diferenţierea dintre fenotipurile de CVID are semnificaţie clinică, întrucât mortalitatea şi morbiditatea variază în grupul cu fenotip al infecţiilor repetate faţă de fenotipul neasociat cu infecţii (fenotipul CVID cu complicaţii neinfecţioase). Astfel, CVID cu complicaţii neinfecţioase tinde să aibă un prognostic mai puţin bun decât CVID cu tiparul clinic al infecţiilor repetate, în ciuda tratamentului substitutiv cu imunoglobuline(8). Deşi clasificarea CVID în funcţie de asocierea sau nu a infecţiilor repetate poate fi limitativă, aceasta poate fi utilă pentru identificarea acelor pacienţi la care este necesar un bilanţ mai extins de investigaţii şi monitorizare postdiagnostică. În grupul pacienţilor cu CVID şi fenotip neinfecţios pot fi avuţi în vedere indivizii cu autoimunitate, boală inflamatorie intestinală, boală pulmonară interstiţială, boală granulomatoasă, afectare hepatică sau neoplazii cunoscute a fi asociate cu CVID, precum limfoamele.

Istoric
Hipogamaglobulinemia
Cu ocazia evaluării iniţiale a pacientului cu suspiciune de imunodeficienţă primară, este esenţială excluderea cauzelor de hipogamaglobulinemie secundară (tabelul 3)(6).
Infecţiile recurente
Infecţiile recurente, mai cu seamă cele ale tractului respirator superior şi inferior (sinuzite, bronşite, pneumonii), constituie cea mai frecventă formă de manifestare a CVID. Cel mai adesea sunt incriminaţi agenţi bacterieni (Streptoccocus pneumoniae, Haemphilus influenzae), însă şi infecţiile virale sunt o cauză semnificativă a infecţiilor recurente ale tractului respirator la aceşti pacienţi. Infecţiile tractului digestiv, exprimate prin sindrom diareic, cauzate de Giardia, Salmonella, Campylobacter ş.a., sunt adesea întâlnite la pacienţii cu CVID. La pacienţii cu fenotipul complicaţiilor neinfecţioase, investigaţiile sunt ghidate de trăsăturile şi/sau complicaţiile bolii, de asocierea afecţiunilor autoimune şi autoinflamatorii(9,10).
Autoimunitatea
Autoimunitatea este cea mai frecventă complicaţie neinfecţioasă a CVID, întâlnită la 5-20% din cazuri. Aceasta este cel mai adesea exprimată prin citopenii autoimune, respectiv trombocitopenie autoimună, anemie hemolitică autoimună sau, mai puţin frecvent, neutropenie autoimună. Manifestările autoimune cu afectarea tractului gastrointestinal includ anemia pernicioasă, gastrita atrofică şi enteropatia autoimună. Manifestările cutanate pot fi reprezentate de alopecie, lichen plan, vitiligo şi psoriazis. Afectarea endocrină autoimună cuprinde endocrinopatiile autoimune, de la hipotiroidism la tireotoxicoză, precum şi diabetul zaharat insulino-necesitant. Alte complicaţii autoimune mai rar întâlnite sunt lupusul eritematos sistemic, vasculita, sindromul antifosfolipidic, scleroza multiplă, miastenia gravis, pancreatita autoimună şi ulcerele orale severe(11,12).
Astfel, diagnosticul de CVID şi documentarea hipogamaglobulinemiei trebuie luate în considerare la pacienţii cu autoimunitate, mai cu seamă la cei care asociază infecţii recurente şi/sau citopenii autoimune.
Boala pulmonară cronică
Este o complicaţie frecventă a CVID, fiind întâlnită la cel puţin 30% dintre pacienţi. Bronşectaziile sunt o complicaţie majoră la aceşti pacienţi. Pneumopatiile interstiţiale sunt întâlnite la 10-20% dintre pacienţii cu CVID şi comportă morbiditate şi mortalitate semnificative. Asocierea frecventă a bolii interstiţiale de plămân cu autoimunitate şi splenomegalie sugerează implicarea anomaliilor de reglare a răspunsului imun şi încadrarea acesteia în fenotipul CVID neasociat cu infecţii(13-15).
Enteropatia
Afectarea digestivă apare la cel puţin 15% din pacienţii cu CVID şi cuprinde infecţii recurente (Giardia, Campylobacter) şi până la afectare autoimună sau neoplazii ori limfoame gastrointestinale. În registrul enteropatiei asociate CVID sunt incluse boli inflamatorii intestinale – boală Crohn, rectocolită ulcerativă, diaree cronică de etiologie neprecizată, hemoragie digestivă, diverticulită, intestin iritabil, esofagită(16-18).
Afectarea hepatică
Boala cronică de ficat este întâlnită la aproximativ 12% din pacienţii cu CVID şi cuprinde granuloame intraparenchimatoase, ciroză biliară primitivă, ciroză, colangită sclerozantă, hiperplazie nodulară regenerativă, hipertensiune portală, varice esofagiene şi hemoragie digestivă, ascită, peritonită bacteriană spontană, sindrom hepatopulmonar şi encefalopatie hepatică. Nu există un consens privind evaluarea în sensul unei imunodeficienţe a pacienţilor cu boală hepatică cronică. De altfel, hipogamaglobulinemia poate surveni în contextul bolii de ficat, ceea ce face dificilă interpretarea corectă a acesteia. Granuloamele intrahepatice şi hiperplazia nodulară regenerativă a ficatului sunt rar întâlnite în afara unei imunodeficienţe primare, astfel încât evaluarea acestor pacienţi în sensul CVID este justificată(19,20).
Hiperplazia limfoidă şi neoplaziile
Sindromul ganglionar şi/sau splenomegalia pot fi trăsături benigne ale CVID sau pot indica sindromul limfoproliferativ şi riscul de evoluţie către limfom. Riscul de dezvoltare a limfomului la pacienţii cu CVID, cel mai adesea non-Hodgkin, este mai mare decât cel al populaţiei generale, mai cu seamă la fenotipul CVID cu complicaţii neinfecţioase. Uneori este imposibil de făcut diferenţierea între CVID complicat cu limfom şi imunodeficienţa umorală secundară terapiei limfomului(8,11).
Istoricul familial
În mod tipic, CVID nu are agregare familială, însă anumite mutaţii care să explice etiologia CVID pot fi suspectate odată cu obţinerea istoricului familial. Mutaţii monogenice, precum cele ale genelor care codifică CTLA4 sau LRBA, ar trebui totuşi avute în vedere în familiile în care o generaţie nu prezintă boala sau când membrii familiei au afectări diferite. S-a demonstrat că variantele genetice identificate la pacienţii cu CVID au penetranţă incompletă şi sunt exprimate prin fenotipuri clinice diferite în rândurile aceleiaşi familii(21).
Evaluarea paraclinică a pacientului cu CVID
Evaluarea de laborator
Evaluarea iniţială cuprinde:
1. Nivelul seric al claselor de imunoglobulină este o evaluare esenţială pentru diagnosticul de CVID, aşa cum s-a discutat în secţiunea dedicată criteriilor de diagnostic. În plus, poate fi avută în vedere evaluarea în dinamică a nivelului seric al IgM. Nivelul crescut al IgM la un pacient cu CVID poate fi semn de boală limfoproliferativă şi pare că asociază o rată mai scăzută a supravieţuirii. Creşterea nivelului IgM serice cu 10 mg/dL sau mai mult în decursul a 20 de luni este asociată cu progresia bolii interstiţiale de plămân, definită prin declinul capacităţii vitale forţate cu 10% sau a DLCO cu 15% din valorile prezise. Studiile au demonstrat că escaladarea nivelului seric al IgM se corelează cu numărul foliculilor limfoizi ectopici de la nivel pulmonar. Astfel, măsurarea în dinamică a nivelului seric al IgM la pacienţii cu CVID şi complicaţii neinfecţioase este recomandată la cei cu hiperplazie LB. S-a observat că ameliorarea şi recrudescenţa bolii interstiţiale de plămân după tratamentul cu rituximab se corelează cu declinul, respectiv creşterea nivelului seric al IgM(7,22).
2. Testele adiţionale de laborator sunt ghidate de tipul afectării şi includ hemoleucograma completă, panelul metabolic de bază – glicemie, calciu, sodiu, potasiu, clor, bicarbonat, albumină serică, proteine totale, probe renale şi hepatice, alături de examenul sumar de urină. Acestea sunt utile ca evaluare generală a afectării prin CVID şi sunt în acelaşi timp utile pentru excluderea unor cauze secundare de hipogamaglobulinemie (tabelul 3). Electroforeza proteinelor serice şi urinare este utilă la pacienţii cu suspiciune de mielom şi ajută astfel la excluderea unei hipogamaglobulinemii în acest context. Hemoleucograma este utilă pentru identificarea citopeniilor, respectiv a trombocitopeniei – o trăsătură frecventă a CVID şi a limfopeniei –, adesea prezentă la pacienţii cu CVID. Un nivel crescut al monocitelor serice (în număr absolut) poate fi documentat la pacienţii cu CVID neasociat cu fenotipul infecţiilor recurente(5).
3. Imunofenotiparea limfocitară poate fi utilă pentru documentarea unor anomalii, precum: nivel redus al limfocitelor B cu memorie (CD19+, CD27+, IgM-, IgD-), tipice CVID cu fenotip cu complicaţii neinfecţioase şi mai rar la CVID cu fenotip al infecţiilor recurente (tabelul 4). CVID este o imunodeficienţă predominant umorală, însă aceasta asociază adesea (în aproximativ jumătate din cazuri) defecte ale limfocitelor T. Aceste defecte pot fi documentate prin intermediul imunofenotipării limfocitare – şi anume, limfopenie T, a subseturilor D4+ şi/sau CD8+. Majoritatea clinicienilor vor clasifica pacienţii cu limfopenie T profundă (LT totale sub 500/µL sau LT CD4+ peste 200/µL) drept pacienţi cu imunodeficienţă combinată, şi nu cu CVID. Fenotipul CVID şi al complicaţiilor neinfecţioase asociază adesea reducerea LT naive CD45 RA+ şi creşterea LT CD45RO+ efectoare şi cu memorie(5).
4. Răspunsul la vaccinare este un test esenţial în diagnosticul CVID. Testele cele mai folosite sunt faţă de toxoidul tetanic, difterie, Haemophilus influenzae şi pneumococ. Vaccinarea cu agenţi vii este contraindicată la pacienţii cu imunodeficienţă! Vaccinul antipneumococic pentru 23 de tulpini este utilizat pentru evaluarea anticorpilor independenţi de LT (tabelul 5)(5,6,7).
Evaluarea gastrointestinală
Testele recomandate la pacienţii cu CVID şi afectare digestivă sunt calprotectina fecală, alfa-1 antitripsina fecală şi nivelul seric al vitaminei B12 (scăzut la pacienţii cu malabsorbţie). Calprotectina fecală este mai sensibilă decât alfa-1 antitripsina în detectarea inflamaţiei mucoasei intestinale şi în monitorizarea activităţii bolii la cei cu boală inflamatorie intestinală. Endoscopia digestivă superioară şi cea inferioară sunt recomandate în screeningul pacienţilor cu CVID, din cauza riscului de neoplazii la sediul gastrointestinal. Studiile au arătat că screeningul endoscopic digestiv în CVID arată modificări la cel puţin 80% dintre pacienţi, respectiv metaplazie intestinală, gastrită atrofică, atrofie vilozitară duodenală sau limfocitoză intraepitelială(5).
Evaluarea pulmonară
ICON recomandă efectuarea unei tomografii computerizate pulmonare şi evaluarea funcţiei pulmonare cât mai aproape de momentul diagnosticului şi, acolo unde se identifică anomalii, monitorizarea prin spirometrie cel puţin o dată pe an. Dispneea este cel mai frecvent simptom al CVID şi al bolii interstiţiale pulmonare(5).
Evaluarea hepatică şi splenică
Testele de laborator nu sunt specifice diagnosticului, astfel încât investigaţii suplimentare, ultrasonografie, CT, RMN, elastografie sau puncţie-biopsie pot fi efectuate, după caz. Splenomegalia este întâlnită la 26-38% din cazurile de CVID. Aceasta poate fi asimptomatică sau poate fi asociată hepatomegaliei şi bolii cronice de ficat. Evaluarea cel puţin anuală a splenomegaliei/ratei de creştere este recomandată. Splenectomia de rutină nu este recomandată, din cauza riscului mare de infecţie, însă poate fi considerată în cazurile severe. Pacienţii cu CVID cu splenomegalie semnificativă tind să asocieze citopenii autoimune sau alte tipuri de limfoproliferare, inclusiv boală pulmonară interstiţială(5).
Evaluarea sindromului ganglionar
Sindromul ganglionar este frecvent întâlnit la pacienţii cu CVID şi, mai puţin în cazurile de limfom, este adesea benign. În cazurile cu evoluţie benignă, biopsia de ganglion limfatic poate arăta hiperplazie limfoidă atipică, reactivă sau inflamaţie granulomatoasă fără infiltrat plasmocitar. Cu toate acestea, în faţa riscului de evoluţie către boală limfoproliferativă, sindromul ganglionar la pacientul cu CVID trebuie monitorizat cu atenţie. Studiile arată un risc de 2,5 ori mai mare de limfom la pacienţii cu CVID şi trăsături limfoproliferative, comparativ cu CVID fără limfoproliferare. În majoritatea cazurilor, limfoamele au sedii extraganglionare şi sunt descoperite la nivel pulmonar sau în ţesuturile asociate mucoaselor(5).
Testele genetice
Testarea genetică permite abordarea terapeutică ţintită atunci când se identifică anumite mutaţii cauzatoare de boală. Deşi în multe cazuri CVID este poligenic şi multifactorial, se estimează că CVID este monogenic în 15-30% din cazuri. O serie de mutaţii genetice care pot fi obiectivate la pacienţii cu CVID sau cu simptome CVID-like ghidează către tratamentul biologic ţintit (tabelul 4)(23,24).
Perspective în evaluarea CVID
Studii în curs evaluează valoarea nivelului seric al antigenului de maturare a LB (BCMA; B Cell Maturation Antigen) ca test diagnostic al CVID şi XLA. Identificarea unui biomarker cu rol diagnostic este necesară, mai cu seamă în situaţia frecventă a cazurilor cu suspiciune de imunodeficienţă primară la care s-a iniţiat deja terapia substitutivă cu imunoglobuline (situaţie în care nivelul IgG seric şi răspunsul la vaccinare nu sunt elocvente, aşa încât, pentru tranşarea diagnosticului în aceste cazuri, se impune sistarea tratamentului pentru cel puţin o lună, ceea ce aduce riscul de infecţii), precum şi la pacienţii cu terapii imunosupresoare, la care hipogamaglobulinemia este dificil de interpretat (persistentă, în contextul unei imunodeficienţe primare versus tranzitorii, secundară tratamentului).
BCMA este un receptor al superfamiliei factorilor de necroză tumorală pentru două citokine: APRIL (proliferation inducing ligand) şi BAFF (B Cell Activating Factor), care promovează activarea şi supravieţuirea LB. BCMA este exprimat aproape exclusiv de către plasmocite. Plasmocitele sunt absente sau reduse semnificativ în CVID. Acestea sunt conţinute predominant la nivelul măduvei osoase şi la nivelul mucoaselor, astfel că pot fi evaluate doar cu ajutorul biopsiei. Întrucât BCMA este clivat endogen de la suprafaţa celulei (plasmocitului) de către o γ-secretază în formă solubilă care poate fi cuantificată la nivel seric, acesta poate fi un test-surogat al măsurării plasmocitelor. BCMA este scăzut în CVID şi în boala Bruton, astfel că studii în curs investighează rolul acestuia ca marker diagnostic(5,25).
Evaluarea cu scop diagnostic şi postdiagnostică a pacientului cu CVID este rezumată în figura 2(5).
Concluzii
CVID este o imunodeficienţă primară cu expresie variabilă, ceea ce denotă nu doar eterogenitatea tabloului clinic, ci şi eterogenitatea anomaliilor imunologice, respectiv afectarea semnificativă a imunităţii umorale şi a producţiei de anticorpi, alături de alterarea variabilă a numărului/funcţiei limfocitelor T. CVID îmbracă în mod clasic fenotipul infecţiilor severe, recurente, însă ultimii ani au permis caracterizarea fenotipului CVID cu complicaţii neinfecţioase. Acesta din urmă asociază un tipar evolutiv mai sever şi o rată de supravieţuire mai redusă. Evaluarea pacientului cu CVID este ghidată de profilul de boală, în CVID cu complicaţii neinfecţioase fiind justificată o evaluare mai extinsă.




Conflict de interese: niciunul declarat
Suport financiar: niciunul declarat
Acest articol este accesibil online, fără taxă, fiind publicat sub licenţa CC-BY.

Bibliografie
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, Klein C, Morio T, Oksenhendler E, Picard C, Puel A, Puck J, Seppänen MRJ, Somech R, Su HC, Sullivan KE, Torgerson TR, Meyts I. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022 Oct;42(7):1473-1507. doi: 10.1007/s10875-022-01289-3. .
- Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, Ochs HD, Oksenhendler E, Puck J, Torgerson TR, Casanova JL, Sullivan KE, Tangye SG. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol. 2020 Jan;40(1):66-81.
- Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova JL, Abel L. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. 2013;33(1):1–7. doi: 10.1007/s10875-012-9751-7.
- Tangye SG, Al-Herz W, Bousfiha A,Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24-64. doi:10.1007/s10875-019-00737-x.
- Lee TK, Gereige JD, Maglione PJ. State-of-the-art diagnostic evaluation of common variable immunodeficiency. Ann Allergy Asthma Immunol. 2021;127(1):19-27. doi: 10.1016/j.anai.2021.03.005. .
- Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract. 2016;4(1):38-59 . doi: 10.1016/j.jaip.2015.07.025.
- Edgar D, Ehl S. ESID Registry – Working definitions for clinical diagnosis of PID. ESID Regist – Work Defin Clin Diagnosis PID. Published online 2016.
- Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650-1657. doi: 10.1182/blood-2011-09-377945.
- Pulvirenti F, Camilli R, Giufrè M, et al. Risk factors for Haemophilus influenzae and pneumococcal respiratory tract colonization in CVID. J Allergy Clin Immunol. 2018;142(6):1999-2002.e3. doi: 10.1016/j.jaci.2018.08.014.
- Oksenhendler E, Gérard L, Fieschi C, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46(10):1547-1554. doi: 10.1086/587669.
- Ho HE, Cunningham-Rundles C. Non-infectious Complications of Common Variable Immunodeficiency: Updated Clinical Spectrum, Sequelae, and Insights to Pathogenesis. Front Immunol. 2020;11:149. doi: 10.3389/fimmu.2020.00149.
- Gereige JD, Maglione PJ. Current Understanding and Recent Developments in Common Variable Immunodeficiency Associated Autoimmunity. Front Immunol. 2019;10:2753. doi: 10.3389/fimmu.2019.0275.3
- Maglione PJ. Chronic Lung Disease in Primary Antibody Deficiency: Diagnosis and Management. Immunol Allergy Clin North Am. 2020;40(3):437-459. doi: 10.1016/j.iac.2020.03.003.
- Weinberger T, Fuleihan R, Cunningham-Rundles C, Maglione PJ. Factors Beyond Lack of Antibody Govern Pulmonary Complications in Primary Antibody Deficiency. J Clin Immunol. 2019;39(4):440-447. doi: 10.1007/s10875-019-00640-.5
- Schussler E, Beasley MB, Maglione PJ. Lung Disease in Primary Antibody Deficiencies. J Allergy Clin Immunol Pract. 2016;4(6):1039-1052. doi: 10.1016/j.jaip.2016.08.005.
- Cunningham-Rundles C, Siegal FP, Cunningham-Rundles S, Lieberman P. Incidence of cancer in 98 patients with common varied immunodeficiency. J Clin Immunol. 1987;7(4):294-299 doi: 10.1007/BF00915550.
- Kinlen LJ, Webster ADB, Bird AG, et al. Prospective study of cancer in patients with hypogammaglobulinaemia. Lancet. 1985;1(8423):263-266. doi: 10.1016/S0140-6736(85)91037-2.
- Mellemkjær L, Hammarström L, Andersen V, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: A combined Danish and Swedish study. Clin Exp Immunol. 2002;130(3):495-500. doi: 10.1046/j.1365-2249.2002.02004.x.
- Fuss IJ, Friend J, Yang Z, et al. Nodular regenerative hyperplasia in Common Variable Immunodeficiency. J Clin Immunol. 2013;33(4):748-758. doi: 10.1007/s10875-013-9873-6.
- Song J, Lleo A, Yang GX, et al. Common Variable Immunodeficiency and Liver Involvement. Clin Rev Allergy Immunol. 2018;55(3):340-351. doi: 10.1007/s12016-017-8638-z.
- Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: One diagnosis to rule them all? J Med Genet. 2016;53(9):575-590. doi: 10.1136/jmedgenet-2015-103690.
- Maglione PJ, Gyimesi G, Cols M, et al. BAFF-driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI insight. 2019;4(5):e122728. doi: 10.1172/jci.insight.122728.
- de Valles-Ibáñnez G, Esteve-Solé A, Piquer M, et al. Evaluating the genetics of common variable immunodeficiency: Monogenetic model and beyond. Front Immunol. 2018;9:636. doi: 10.3389/fimmu.2018.00636.
- Maffucci P, Filion CA, Boisson B, et al. Genetic diagnosis using whole exomesequencing in common variable Front Immunol. 2016;7:220. doi: 10.3389/fimmu.2016.00220.
- Years of Potential Life Lost due to Cancer – United States, 1968–1985. JAMA . 1989;261(2):209 doi: 10.1001/jama.1989.03420020053013.

Angioedemul ereditar cu deficienţă de C1-inhibitor esterază. Particularităţi la populaţia pediatrică
Noémi-Anna Bara, Réka Borka-Balás
Angioedemul ereditar (AEE) cu deficienţă de C1-inhibitor esterază (C1-INH ) este o boală genetică rară, care are o prevalenţă de 1:50000 de locuitori(1,12). Deficienţa cantitativă (HAE tip 1) sau disf...
Recomandări cu privire la vaccinarea copiilor alergici
Imunizarea copiilor este o măsură indispensabilă de sănătate publică, eficientă în prevenţia bolilor infecţioase. Pacienţii alergici sunt încurajaţi să practice aceleaşi măsuri publice de imunizare ca şi cei nonalergici, cu excepţia cazurilor foarte rare în care riscurile asociate vaccinării depăşesc benefici...
Cauzele imunologice în avortul spontan recurent
Cristina Uţa, Carmen Panaitescu
Avortul spontan recurent (ASR) afectează 1-3% dintre cupluri, fiind definit prin trei pierderi consecutive de sarcină înainte de 20 de săptămâni de gestaţie (Günther et al., 2018). Conform Societăţii Americane pentru Reproducere (American Society for Reproductive Medicine (ASRM), ASR poate fi diagnosticat la ...
Recomandări cu privire la vaccinarea copiilor alergici
Imunizarea copiilor este o măsură indispensabilă de sănătate publică, eficientă în prevenţia bolilor infecţioase. Pacienţii alergici sunt încurajaţi să practice aceleaşi măsuri publice de imunizare ca şi cei nonalergici, cu excepţia cazurilor foarte rare în care riscurile asociate vaccinării depăşesc benefici...
Cauzele imunologice în avortul spontan recurent
Cristina Uţa, Carmen Panaitescu
Avortul spontan recurent (ASR) afectează 1-3% dintre cupluri, fiind definit prin trei pierderi consecutive de sarcină înainte de 20 de săptămâni de gestaţie (Günther et al., 2018). Conform Societăţii Americane pentru Reproducere (American Society for Reproductive Medicine (ASRM), ASR poate fi diagnosticat la ...