Holoprosencephaly is a brain abnormality resulting from an incomplete cleavage of the prosencephalon during early embryogenesis. Uncontrolled maternal diabetes mellitus is one of the strongest human teratogens and the most well-known cause of holoprosencephaly. We report a rare case of holoprosencephaly in a male fetus in a preexisting diabetic pregnancy. The diagnosis of the holoprosencephaly was established by ultrasound scan and MRI data.
OBSTETRICS
Lobar holoprosencephaly associated with maternal diabetes mellitus – case report
Holoprozencefalie lobară asociată diabetului zaharat matern – prezentare de caz
First published: 28 martie 2024
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Gine.43.1.2024.9419
Abstract
Rezumat
Holoprozencefalia este o anomalie a creierului cauzată de un clivaj incomplet al prozencefalului în timpul embriogenezei timpurii. Diabetul zaharat matern necontrolat este unul dintre cei mai puternici agenţi teratogeni şi cea mai frecventă cauză de holoprozencefalie. Raportăm un caz rar de holoprozencefalie la un făt de sex masculin într-o sarcină complicată cu diabet zaharat pregestaţional. Diagnosticul de holoprozencefalie a fost stabilit prin ecografie şi evaluare IRM.
Introduction
The term holoprosencephaly (HPE) describes a spectrum of cerebral and facial malformations that result from an absent or incomplete division of embryonic forebrain, the prosencephalon. It is a condition that occurs in the first two or three weeks of pregnancy. The lack of division results in a complete or partial union of the forebrain and in a partial communication or in a single ventricle instead of two lateral ventricles(1). This is the most common brain malformation, with an incidence of 1:250 during prenatal life and 1:6000 among live births(2,3).
Holoprosencephaly results if the prosencephalon does not properly segment or if it does not segment completely. There are three main types of holoprosencephaly according to severity: alobar – no segmentation, the most severe form, and semi-lobar and lobar, characterized by partial segmentation. Lobar holoprosencephaly is the least severe of the classical subtypes of holoprosencephaly(4).
Recently, a fourth subtype of holoprosencephaly was described, known as middle interhemispheric variant (MIH; syntelencephaly), the least severe form of holoprosencephaly, characterized by the almost complete segmentation of prosencephalon(5). Other conditions sometimes included in the spectrum of holoprosencephaly include septo-optic dysplasia, which is associated with subtle craniofacial malformations and mild developmental delay(6,7). The prognosis depends on the type of holoprosencephaly. Generally, in the alobar type, the baby is either stillborn or dies soon after birth, therefore alobar and semilobar forms are usually lethal within the first year of life. In lobar holoprosencephaly, life expectancy may be normal but, usually, with severe developmental delay and visual impairment(4-6).
The most-well known cause of holoprosencephaly in humans is maternal type 2 diabetes mellitus. Maternal diabetes is the only confirmed human teratogen associated with this condition. The chance for holoprosencephaly to occur in pregnancies of diabetic mothers is about 1%, being 200-fold increased as compared with controls(5,6). Pregestational diabetes mellitus increases the risk for structural birth defects affecting both maternal and neonatal pregnancy outcome. Prenatal exposure to hyperglycemia can result in spontaneous abortions, perinatal mortality and malformations. The most common birth defects associated with diabetes mellitus are congenital heart defects and central nervous system defects(6).
We report a case that could be defined as a lobar form of holoprosencephaly in a diabetic mother.
Case report
We report the case of a 30-year-old primigravida presented to the Department of Obstetrics and Gynecology for a routine nuchal ultrasound examination. There wasn’t a history of marital consanguinity. Ultrasonography revealed a single live intrauterine fetus of 12 weeks. The patient was type 2 diabetic and had chronic hypertension. She was on nifedipine, insulin, metformin, folic acid and prenatal vitamins. There was no family history of holoprosencephaly. Serology assessment eliminated recent toxoplasmosis or rubella infection. The brain views were abnormal. The cerebral hemispheres were separated both anteriorly and posteriorly, but there was communication in the anterior horns of the lateral ventricles. There was a wide communication of this fused segment with the third ventricle. The ultrasound findings were consistent with lobar holoprosencephaly. The rest of the fetal anatomy appeared normal. Amniocentesis revealed normal genetic component (PCR + array-CGH). Whole exome sequencing also demonstrated normal results. Fetal brain MRI at 21 weeks showed a middle interhemispheric variant of holoprosencephaly with some parietal frontal lobe fusion. At 23 weeks, fetal growth restriction with abnormal Dopplers and severe oligohydramnios without history of ruptured membranes were noted. After discussion, due to risk of preeclampsia and the substantial risk of handicap caused by holoprosencephaly, prematurity and severe fetal growth restriction, the couple decided to end the pregnancy. Pregnancy termination was performed at 23 weeks, with a vaginal delivery of a male fetus.
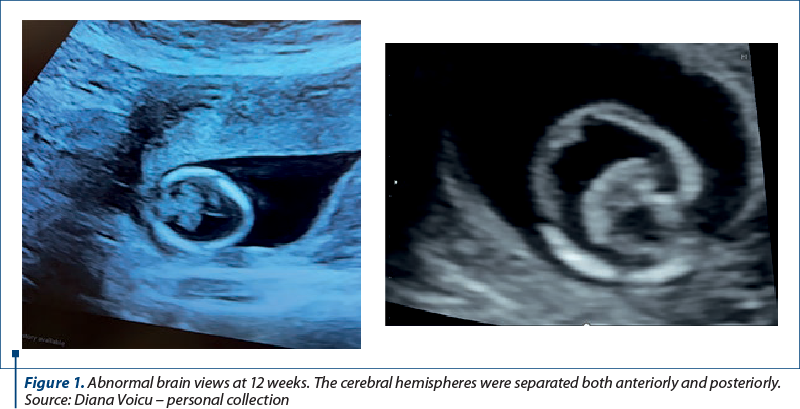
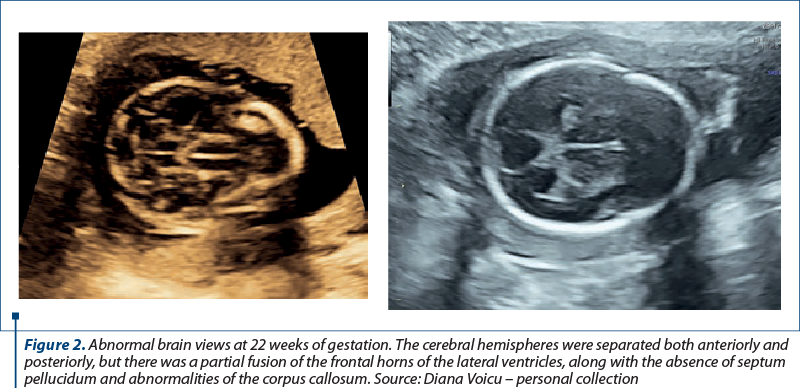
Discussion
In 1983, Barr Jr. et al. reported seven infants of diabetic mothers affected by holoprosencephaly. The seven cases of holoprosencephaly in infants of diabetic mothers were noted over four years at three medical centers. The diagnosis of HPE was confirmed by autopsy in four cases and by computed tomography in two cases. One infant had facial features characteristics of HPE. In five out of seven patients, an isolated malformation sequence was present, consisting of HPE and its variable effects on development of the face and brain(5,6).
At the University of Iowa, 19 malformed infants were noted after review of medical records of 105 infants of diabetic mothers delivered from 1962 to 1976. Seven infants had central nervous system defects, and one of these had agenesis of the corpus callosum, fusion of the frontal lobes and a common lateral ventricle. At the University of Michigan, nine infants with malformations were identified retrospectively among 84 infants of diabetic mothers born from 1967 to 1979(8-10).
Holoprosencephaly is highly lethal during fetal life. It has been estimated that only 3% of HPE fetuses are liveborn. The condition is a profound fetal brain anomaly that cannot be altered or treated. Considerations for management of pregnancy include elective termination if the diagnosis is made earlier than 24 weeks. Because 30-50% of fetuses with HPE have chromosomal abnormalities, prenatal karyotype is strongly recommended. Trisomy 13 accounts for 75% of the chromosomal abnormalities. Since familial recurrences of HPE was reported, a family history should be obtained(7,8).
Conclusions
Pregestational diabetes is a rising problem, with an important impact on adverse pregnancy outcomes(9,10).
Holoprosencephaly, like all fetal defects, should be suspected in malformed infants of diabetic mothers(11). These defects should also be specifically looked for during the ultrasonographic examination of the fetus of the insulin-dependent diabetic mothers, particularly when diabetes control during the early weeks of pregnancy has not been optimal(12-14).
Corresponding author:
Diana-Ioana Voicu
E-mail: voicu_diana06@yahoo.com
Conflict of interest: none declared.
financial support: none declared.
This work is permanently accessible online free of charge and published under the CC-BY licence.

Bibliografie
- Abe Y, Kruszka P, Martinez AF, Roessler E, Shiota K, Yamada S, Muenke M. Clinical and demographic evaluation of a holoprosencephaly cohort from the Kyoto collection of human embryos. Anatl Rec (Hoboken). 2018;301(6):973-86.
- Kauvar EF, Muenke M. Holoprosencephaly: recommendations for diagnosis and management. Curr Opin Pediatr. 2010;22(6):687-95.
- Kruszka P, Muenke M. Syndromes associated with holoprosencephaly. Am J Med Genet C Semin Med Genet. 2018;178(2):229-37.
- Winter TC, Kennedy AM, Woodward PJ. Holoprosencephaly: a survey of the entity, with embryology and fetal imaging. Radiographics. 2015;35(1):275-90.
- Barr M Jr, Hanson JW, Currey K, Sharp S, et al. Holoprosencephaly in infants of diabetic mothers. J Pediatr. 1983;102(4):565-8.
- Reece EA, Homko CJ, Wu YK. Diabetic embryopathy. Fetal Maternal Med Rev. 1996;8(4):187-97.
- Matsunaga EI, Shiota K. Holoprosencephaly in human embryos: epidemiologic studies of 150 cases. Teratology. 1977;16(3):261-72.
- Passarge E, Lenz W. Syndrome of caudal regression in infants of diabetic mothers: Observations of further cases. Pediatrics. 1966;37(4):672.
- Myrianthopoulos NC, Chung CS. Congenital malformations in singletons: Epidemiologic survey. Birth Defects Orig Artic Ser. 1974;10(11):24.
- Roach E, Demyer W, ConneaHy PM, Palmer C, Merrit AD. Holoprosencephaly: Birth data, genetic and demographic analyses of 30 families. Birth Defects Orig Artic Ser. 1975;11(2):294-313.
- Reece EA, Homko CJ, Wu YK. Multifactorial basis of the syndrome of diabetic embryopathy. Teratology. 1996;54(4):171-82.
- Kousseff BG. Diabetic embryopathy. Curr Opin Pediatr. 1999;11(4):348-52.
- Wentzel P, Eriksson UJ. Embryopathy and diabetes. In: Lapolla A, Metzger BE (Ed). Gestational diabetes: a decade after the HAPO Study. Karger Publishers, Basel, Switzerland. 2020;28:132-44.
- Ming JE, Muenke M. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am J Human Genetics. 2002;71(5):1017-32.