This article aims at presenting the modern tendencies to redefine the role of iron into the normal and pathological metabolic processes, reminding the normal metabolic pathways of iron, and then compare the normal findings with pathological ones, mostly in cancers. One relatively new concept is “ferroptosis”, a different type of (cancer) death cell, intensely researched, at least in the past 11 years. All of these results could potentially have an impact in the better understanding of the metabolism of cancer cells and in the therapeutic area, as a practical ultimate endpoint.
REVIEW
Iron, ferroptosis and association with tumoral evolution and potential therapeutic impact
Fierul, feroptoza şi asocierea cu evoluţia tumorală şi potenţialul impact terapeutic
First published: 23 martie 2023
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/OnHe.62.1.2023.7744
Abstract
Rezumat
Articolul îşi propune să arate tendinţele moderne de redefinire a rolului fierului în procesele metabolice normale şi patologice, reamintind căile metabolice normale ale fierului, apoi compararea elementelor normale cu cele patologice, mai ales în cazul cancerelor. Un concept relativ nou este cel denumit „feroptoză”, un mod diferit de moarte celulară (a celulelor canceroase), cercetat intens cel puţin în ultimii 11 ani. Toate aceste rezultate pot avea un impact în înţelegerea mai bună a metabolismului celulelor canceroase şi, ca obiectiv final, în aria terapeutică.
Introduction
Iron is essential for the oxidation-reduction reaction and can exhibit a wide range of oxidation states, which makes it a multifunctional participant in redox reactions. Iron plays a vital role in various cellular processes, such as cellular respiration (e.g., cytochrome c oxidase, ferredoxin, cytochrome, and Rieske protein), energy metabolism (e.g., aconitase, citrate synthase, succinate dehydrogenase and isocitrate dehydrogenase), DNA replication, DNA synthesis and nucleic acid repair (e.g., the catalytic subunit of replicative DNA polymerases, DNA helicase and ribonucleotide reductase), and iron-dependent signaling(1).
Iron is also used in the synthesis of heme and iron-sulfur clusters (ISC), which are incorporated into proteins that carry out the citric acid cycle, oxidative phosphorylation, and many other essential functions(1,9).
Iron is essential for the normal physiological function of the human body, but it may also be toxic, as it generates a large number of free radicals in the presence of hydrogen peroxide. For example, in the Fenton reaction, ferrous iron (Fe2+) reacts with hydrogen peroxide to be oxidized to ferric iron (Fe3+) while generating hydroxyl radicals. When superoxide is present, the Fe3+ produced by the Fenton reaction can be reduced to Fe2+, and then the Fenton reaction will proceed again, which is called the Haber-Weiss reaction. Both the Fenton reaction and the Haber-Weiss reaction can produce a large number of hydroxyl radicals – one of the most important oxidants found in human body, which can lead to peroxidation and apoptosis by attacking protein, lipids, nucleic acids and carbohydrates. In addition, excessive free radicals in the human body can lead to cell and tissue organ damage, and these processes are closely related to tumorigenesis(1,2,9,10).
Physiological mechanisms of iron regulation
Iron is mainly present in the oxidized state (Fe3+) and is divided into dietary iron and environmental iron. Dietary iron primarily exists as either nonheme bound iron, or heme. Heme iron has a higher absorption rather than nonheme bound iron. Iron in the diet is mainly reduced to Fe2+ in the duodenum by duodenal cytochrome B (DCYTB) and absorbed into intestinal epithelial cells under the synergistic effect of divalent metal transporter 1 (DMT1). Heme iron is also absorbed by intestinal cells through an unknown mechanism and is metabolized by heme oxygenase-1 (HO-1) to release Fe2+. Heme iron is absorbed by intestinal cells (unknown mechanism) and degraded by heme oxygenase-1 (HO-1), releasing Fe2+, which is transported out of the cells by the iron efflux pump ferroportin (FPN1) on the basal side of the intestinal epithelial cells, and consequently oxidized into Fe3+ by hephaestin (HEPH); then Fe3+ binds to transferrin (TF) and enters the circulation through the portal vein; here, each transferrin can bind two Fe3+ to form TF- [Fe3+]2 complex, which binds to transferrin receptor (TFR1) on the cell surface and is absorbed into cells to form endosome. Subsequently, it is reduced to Fe2+ by six-transmembrane prostate epithelial antigen 3 (STEAP3), which is then transported into cytoplasm by DMT1 to exert physiological functions or constitute the cytoplasmic labile iron pool (LIP). LIP can be taken up by non-hematopoietic cells, causing parenchymal iron deposition which can result in free radical damage(1).
Intracellular iron homeostasis is mainly regulated through an iron-dependent protein network, including iron-responsive element binding proteins (IRPs), in which IRP1 and IRP2 are important components. Thioredoxin family proteins are important mediators in iron metabolism, since these proteins regulate the expression of IRPs. To ensure iron homeostasis, IRPs binds to the iron responsive element (IRE) on the untranslated region of messenger RNA encoding the protein essential for cellular iron regulation (TFR1, DMT1), thereby participating in iron uptake (TFR1), storage (FT), redistribution, and efflux (FPN1)(1).
When intracellular iron is low, IRPs can inhibit the translation of FPN1 and FT, but increases the protein synthesis of TFR1(1).
In contrast, when the intracellular iron is abundant, the synthesis of FPN1 and FT is increased due to the instability of IRP, while the degradation of TFR1 is promoted. IRP1/2 are key iron regulators for the maintenance of cellular iron homeostasis. IRP1 is a cytosolic aconitase, an enzyme containing a [4Fe-4S] cluster(1,3,7,9).
In the absence of intracellular iron, there is insufficient iron for Fe-S biogenesis, leaving an incomplete [3Fe-4S] cluster. The enzymatic activity of aconitase is lost and this protein then initiates its IRP activity, as IRP1. When the protein contains the [3Fe-4S] cluster, it can bind to IREs. Through these mechanisms, IRPs can meet the metabolic needs of cellular iron and also minimize the toxic effects of excessive iron(1,7,9).
The excess iron is mainly stored in liver, which also acts as an iron-sensing organ and controls systemic iron through the secretion of the peptide hormone hepcidin. Hepcidin is a 25-amino acid short peptide produced by the liver and encoded by the HAMP. FPN1, which is mainly expressed in the cell membrane and cytoplasm, is the only known 571-amino acid transmembrane protein in vertebrates that transports iron from intracellular to extracellular side. When the intracellular or circulating iron level is relatively high, hepcidin is generated from hepatocytes and secreted into the circulation system through a bone morphogenetic protein (BMP6)-mediated pathway. Hepcidin can trigger the internalization and subsequent lysosomal degradation of FPN1 via binding to the basal side of the intestinal epithelium and to the FPN1 on the macrophage surface, preventing iron absorption from the digestive tract and iron circulation from the body, respectively. The reduction of iron modulators causes FPN1 to absorb iron and increases systemic iron levels.
Therefore, FPN1 and ferritin are essential for maintaining iron homeostasis in the body(1,7).
Ferritin is a complex that can hold 4500 Fe3+ ions and is widely present in mammalian cells. The ferritin complex is assembled by the heavy chain (ferritin heavy chain 1; FTH1) and the light chain (ferritin light chain; FTL). FTH1 has a ferrous oxidase activity and can catalyze the oxidation of Fe2+ to Fe3+, reducing a large number of Fe2+-produced free radicals participating in the Fenton reaction, and thus the damage of free radicals to tissues and organs. Iron combines with each other through ferritin iron pores and Fe2+ is further oxidized to Fe3+ by FTH1 in the ferritin cage, resulting in the inert deposition of Fe3+, which cannot use or generate ROS in cells. The main way of ferritin to release iron is selective autophagy mediated by NCOA4, which binds to ferritin and is transported to lysosomes, where ferritin is degraded and iron is released for cell use(1,7,10).
Iron metabolism in cancer
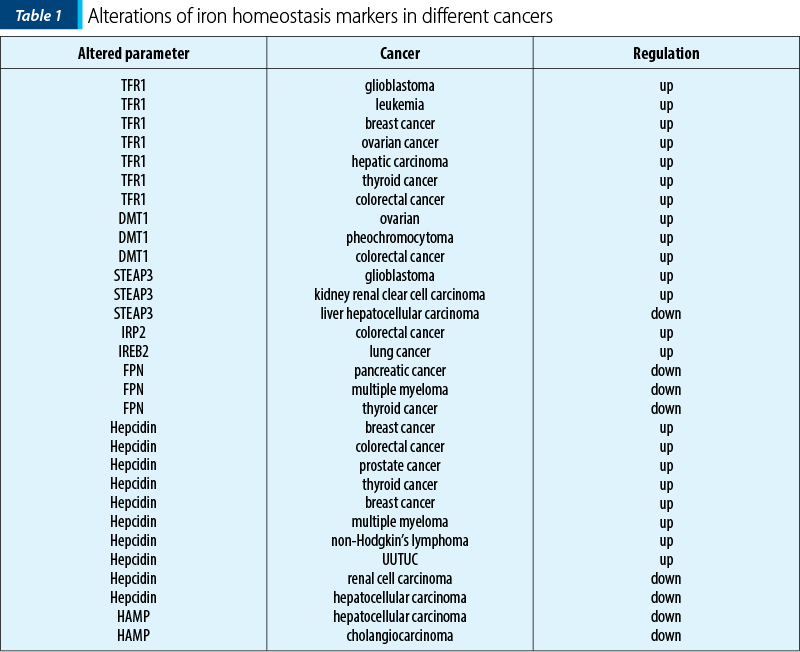
A large number of studies have shown that abnormal iron homeostasis is one of the markers of cancer (Table 1). Tumor cells demand for iron is also significantly higher than that of normal cells (their metabolism and proliferation rate are generally higher than that of normal cells, this leading to the exceeding oxidative stress). For survival, tumor cells can develop a concomitant up-regulation of antioxidant defenses, such as activating antioxidant transcription factors and promoting the expression of various antioxidant genes. And, conversely, since tumor cells are strongly dependent on iron for their growth/proliferation, they are more sensitive to iron depletion than normal cells. This imbalance is mainly manifested in cancer as increased iron metabolism, iron affinity, iron input, and inhibition of its output, thereby completing iron accumulation(1).
Iron uptake(1)
TFR1 is significantly up-regulated in breast cancer. Also, subsequent studies confirmed that TFR1 is highly expressed in various cancers such as glioma, leukemia, breast cancer and ovarian cancer.
Estrogen inhibited the synthesis of ferritin and enhanced the efflux of intracellular iron. Meanwhile, combining with doxorubicin, estrogen significantly reduced TFR1 expression and enhanced the sensitivity of breast cancer cells to doxorubicin.
A higher expression of TFR1 is closely related to the activation of ERK signaling pathway in thyroid cancer, leading to the disorder of genes involved in abnormal accumulation of intracellular free iron and drug resistance.
DMT1 is highly expressed in various types of cancers, such as colorectal cancer and ovarian cancer.
Wang et al. found that DMT1, TFR1 and ferritin were highly expressed in ovarian cancer cells and the overexpression of DMT1 promoted the progression of ovarian tumor.
In SDHB-mutated pheochromocytoma, iron accumulation caused by significant up-regulation of iron transport-related proteins, such as DMT1, TF and TFR2, can increase oxidative stress to some extent.
In colorectal cancer, DMT1 can be induced through hypoxia-inducible factor 2a-dependent transcription. However, in hepatocellular carcinoma, patients with a lower expression of DMT1 displayed a worse disease-free survival, and this effect was more significant in patients with advanced hepatocellular carcinoma.
The carcinogenic activity of DMT1 is tightly correlated with its iron-transport activity, characterized by the evidence that tumor in DMT1-knocked out mice was weakened when being fed with a high-iron diet. Additionally, desferoxamine (DFO) increases iron concentration by up-regulating the expression of DMT1 and TFR1, thereby promoting the migration of breast cancer cells.
Propofol, which is widely used in clinical practice for intraoperative general anesthesia and postoperative sedation, regulates DMT1 expression by modifying Ca2+-permeable a-amino-3-hydroxyl-5-methylisoxazole-4-propionic acid receptors (CPARs), inhibiting tumor oxidative stress and glioma tumor growth.
STEAPs are up-regulated in tumors and they can promote tumor cell proliferation as well as suppress apoptosis. STEAP is a family of prostate-specific cell-surface antigen highly expressed in human prostate tumors. The STEAP gene for six transmembrane epithelial antigen of the prostate is also up-regulated in multiple cancer cell lines, such as blader, colon, glioma, ovarian and Ewing sarcoma.
For example, STEAP3 promotes glioma cell proliferation, invasion and spheroid formation by inducing mesenchymal transformation, promoting TFR1 expression, and activating the STAT3-FoxM1 axis; in colorectal cancer, STEAP3 expression is significantly higher in tumor than that in colonic mucosa.
Similarly, STEAP2 accelerates prostate cancer progression by promoting proliferation, migration and invasion by regulating the transcriptional profiles of some genes involved in metastasis. Burnell et al. proved that reducing the expression of STEAP2 inhibited the proliferation, migration and invasion in prostate cancer cells.
Intracellular iron regulation(1)
IRP1 or IRP2 can increase intracellular iron, and the abnormal activation of them is closely related to many cancers.
IRP2 is overexpressed in colorectal cancer compared with normal colonic mucosa, and is positively correlated with TFR1 expression. In addition, the expression of IRP2 is associated with mutations of BRAF, which primarily occur in the early stages of colorectal cancer and is often associated with poor prognosis.
Notably, chemotherapy and targeted therapy may work together to disrupt IRP-mediated iron regulation. Horniblow et al. found that the MEK inhibitor trimetinib consistently inhibited IRP2 expression in four colorectal cell lines, resulting in decreased TFR1 expression and increased ferritin expression. Miyazawa et al. found that cisplatin bound to Cys512 and Cys516 of human IRP2 and destroyed its function, based on which DFO combined with cisplatin resulted in increased iron consumption and reduced tumor growth in a mouse xenograft model of colonic adenocarcinoma.
Iron efflux in cancer(1)
Ferroportin FPN1, the only iron export protein, is involved in the regulation of intracellular iron concentration, and its abnormal down-regulation is also observed in most tumors.
The suppressive roles of FPN1 have been established in tumors, such as in prostate cancer cell lines, where a low FPN1 level caused by up-regulation of ferritin promoted proliferation, migration and apoptotic resistance, and overexpression of FPN1 induced p53 and autophagy, and reduced tumor growth in vivo. Overexpression of FPN1 decreased proliferation, colony formation and tumor growth, as well as liver metastasis in breast cancer.
Various studies observed that FPN1 regulates tumor progression via destroying iron homeostasis, and a study demonstrated that cadmium (Cd)-induced MDA-MB-231 cell proliferation, EMT and migration were caused by inhibiting FPN1 expression and associated with destruction of iron homeostasis, and hepcidin secreted by thyroid cancer cells could decrease FPN1 and retain intracellular iron, thereby promoting cancer proliferation.
Another study showed that FPN1-mediated iron metabolism might play a role in the chemosensitivity and treatment outcome of acute myeloid leukemia.
Hepcidin disorders of iron modulators in cancer cause changes in iron homeostasis. Hepcidin, a negative regulator of FPN1, is significantly up-regulated in various tumors, such as breast, colorectal and prostate cancer. Numerous studies have shown the promoting effects of hepcidin on tumor progression. Zhao et al. confirmed that hepcidin enhanced the proliferation, migration and anti-apoptotic capabilities of prostate cancer cells by reducing the expression of FPN1 and increasing intracellular iron level.
Serum hepcidin is elevated in many cancer patients, including prostate cancer, breast cancer, multiple myeloma and non-Hodgkin’s lymphoma. High serum hepcidin was associated with cancer recurrence and metastasis.
Strikingly, liver cancers showed a drastic reduction of hepcidin expression compared to benign liver tissues. In Caucasian patients with no history of alcohol consumption, down-regulation of hepcidin is associated with rapid cancer progression and poor disease-specific survival. Hepcidin expression is positively correlated with BMP6/interleukin 6 (IL6) cytokines and cytotoxic immune infiltration in liver cancer tissues.
Heme has been indicated to promote cell proliferation in leukemia and lung cancer, and increase HO-1 activity promotes invasion and migration of breast cancer cells. Up-regulation of BMP, which induces cancer cells to secrete hepcidin, has also been observed in various tumors, including breast, prostate and bladder cancers.
Concluding, cancer cells usually increase the input of iron and inhibit its output, thereby achieving iron accumulation. However, it is not entirely clear how they respond to the increased instability.
Iron and cell death
Ferroptosis is a recently described non-apoptotic programmed cell death mechanism that is characterized by the buildup of iron (Fe)-dependent lipid peroxides in cells, and is morphologically, biochemically and genetically distinct from other forms of cell death, having emerged to play an important role in cancer biology. Dixon suggested ferroptosis as a new mode of cell death for the first time in 2012. More briefly is defined as iron-dependent regulatory necrosis caused by membrane damage mediated by massive lipid peroxidation(1-4).
During ferroptosis, cells show unique features, such as: cell membrane rupture, cytoplasmic swelling, cytoplasmic organelle swelling, mitochondrial membrane density increase, mitochondrial cristae reduction/disappearance, mitochondrial outer membrane rupture etc. It is regulated by multiple cellular metabolic events, including redox homeostasis, iron handling, mitochondrial activity and metabolism of amino acids, lipids and sugars, in addition to numerous signaling pathways relevant to disease(1,3).
Ferroptosis can occur through two main pathways: the external or transporter-dependent pathway, and the internal or enzyme-regulated pathway(1,3,5,6).
Ferroptosis is caused by the redox imbalance between the production of oxidants and antioxidants, which is driven by the abnormal expression and activity of numerous redox active enzymes that produce or detoxify free radicals and lipid oxidation products is defined as the effect of iron-dependent regulatory necrosis caused by membrane damage mediated by massive lipid peroxidation. It is characterized by high intracellular levels of iron and reactive oxygen species (ROS). It is mainly caused by deficits in the production of reduced glutathione or by the dysfunction of Gpx4, which are ROS eliminators(1,2). Excess ROS induce lipid peroxidation and disintegration of lipid membranes, then cell death.
Intriguingly, therapy-resistant cancer cells, particularly those of the mesenchymal state and prone to metastasis, are particularly vulnerable to ferroptosis(3).
Ferroptosis has significant importance during cancer treatment due to a combination of factors, including suppression of the glutathione peroxidase 4 (Gpx4), cysteine deficiency and arachidonoyl (AA) peroxidation, which cause cells to undergo ferroptosis.
However, the physiological significance of ferroptosis throughout development is still not fully understood(1-7).
Additionally, some researchers found that a diet rich in n-3 long-chain unsaturated fatty acids significantly inhibited tumor growth in mice, indicating that the supplementation of dietary unsaturated fatty acids can serve as a selective adjuvant antitumoral approach. Furthermore, supplementation of dietary unsaturated fatty acids may represent a selective adjuvant antitumor approach.
Many extracts from plants and herbs also exhibit antitumor effects by inducing ferroptosis. Wang et al. found that quercetin could promote the degradation of lysosomal-dependent ferritin and the release of free iron, this effect and quercetin-induced ROS production synergistically leading to lipid peroxidation and ferroptosis(1-4).
Overview of recent research progress on ferroptosis. GPX4, BH4, FSP1 and DHODH are the main defense system of ferroptosis. ACSL4 and the Fenton reaction promote ferroptosis, and p53 has dual roles in ferroptosis(2).
Recently, ferroptosis is being more recognized as an adaptive characteristic that helps the body destroy cancerous cells. It plays a critical role in the prevention of tumor formation by eliminating cells that are lacking essential nutrients and water and which have been damaged by infection or environmental stress.
For the moment, there were described the following mechanisms of ferroptosis(2,4-6):
1. Ferroptosis due to suppression of system Xc-. A heteromeric amino acid transporter, system Xc-, is expressed in the plasma membrane of many cell types and is the target of type I ferroptosis activators (erastin, sulfasalazine and sorafenib).
2. Ferroptosis involving Gpx4. Gpx4 has been shown to reduce mitochondrial apoptosis in preliminary studies. This suggests that, whereas Gpx4 is primarily responsible for controlling ferroptosis, its absence in some cell types may also result in other types of cell death. Ferroptosis and other cell death processes are yet unknown. It is unclear whether the subcellular location of lipid peroxidation is important in inducing ferroptosis, or ferroptotic cell death executors are still unidentified.
3. Ferroptosis involving mitochondrial voltage-dependent anions channels. The mitochondrial voltage-dependent anions channels (VDACs) proteins are beta-barrel porins that span the outermost mitochondrial membrane and are responsible for permeability.
4. Some other pathways involving ferroptosis:
Activation of long-chain polyunsaturated fatty acids (PUFAs) is thought to have a role in ferroptosis because these PUFAs may enhance the likelihood of the formation of lipid peroxidation when they are included in phospholipids. However, it is still debated whether this process is mediated by specific lipoxygenase or whether it is caused either by Fe-dependent, radical-mediated Fenton reaction, or autoxidation of lipid bilayers.
Lipid peroxidation inhibitors or efforts to reduce the amount of PUFAs on the membrane surface are the most effective ways to avoid ferroptosis. Similarly, Fe chelation has been shown to protect against oxidative stress and ferroptotic death of cells frequently.
Two independent research groups revealed ferroptosis suppressor protein-1 (FSP-1) – previously known as apoptosis-inducing factor mitochondria associated 2 (AIFM2) – as a novel ferroptosis regulator. FSP-1 was discovered to be a very effective antiferroptotic enzyme, and overexpression of this enzyme resulted in full rescue from ferroptosis produced by chromosomal deletion or pharmacological suppression of Gpx4 (a ferroptotic enzyme). Because of its oxidoreductase activity, FSP-1 has an antiferroptotic effect on extramitochondrial ubiquinone (CoQ10), which it converts to ubiquinol by reducing it with NAD(P)H/H+.
Sulfur transfer pathways may play a role in the incidence of ferroptosis. When exposed to oxidative stress, methionine may be transformed into cystine through the sulfur transfer route, and then GSH can be produced to further enhance the antioxidant properties of the amino acid.
5. Role of mitochondria in ferroptosis. Mitochondrial lipids seem to be key sources of lipid peroxides during ferroptosis.
6. Role of lysosome in ferroptosis. Lysosomes are also involved in the induction of ferroptosis. Lysosome is the primary organelle responsible for the autophagic destruction of protein aggregates and, experimentally, the lysosome is the most important cellular ROS reservoir in HT1080 cells exposed to substances which induce ferroptosis.
7. Role of Fe metabolism in ferroptosis. Ferroptosis is regulated by oncogenes, and the most frequently mutated gene, p53, as noted above, has been proven to regulate ferroptosis in dual manners(2).
Iron and ferritinophagy
Ferritinophagy is a type of cell selective autophagy in which the ferritin (mainly ferritin heavy chain 1) is degraded into autophagosomes mediated by nuclear receptor coactivator 4 (NCOA4), leading to the ferritin-bound iron to be released as free iron. Ferritinophagy contributes to the initiation of ferroptosis through degradation of ferritin, which triggers labile iron overload (IO), lipid peroxidation, membrane damage and cell death, and plays a certain role in tumorigenesis(1).
When cellular iron level is high, NCOA4 and HERC2 have an iron-dependent effect, resulting in the degradation of NCOA4 through ubiquitin proteasome system, thereby reducing the release of iron and facilitating the storage of ferritin. Under low cellular iron conditions, the interaction between NCOA4 and HERC2 is low, leading to an increase of NCOA4 level, increasing the degradation of ferritin and iron autophagy flux to supplement cellular iron(1).
Thus, a proper amount of ferritinophagy can maintain the balance of iron in cells(1).
An activation in excess of ferritinophagy can lead to disease induced by iron overload. Studies have found that there is a close relation between iron release caused by ferritinophagy and ROS damage in ferroptosis. Under certain conditions, iron release caused by ferritinophagy is a component of ferroptosis, and may be a direct driving factor of ferroptosis. The role of ferritinophagy-related genes in cancer progression has been confirmed. Ferritinophagy is considered a form of autophagic ferroptosis. There are many studies focused on clarifying the new discoveries regarding NCOA4 and FTH1 in cancer(1).
Ferroptosis and anticancer drugs
Numerous organ injuries and degenerative pathologies are driven by ferroptosis. As such, pharmacological modulation of ferroptosis, via both its induction and inhibition, holds great potential for the treatment of drug-resistant cancers, ischemic organ injuries and other degenerative diseases linked to massive lipid peroxidation(3,8,9).
Studies have also demonstrated that activating ferroptosis and apoptosis immensely increased chemotherapy sensitivity, which might provide strategies for the combination therapy for cancers(2,3).
Ferroptosis inducers, including 1S, 3R-RSL3 (RSL3), which inhibits the function of GPX4 and erastin, which decreases the level of reduced glutathion via the inhibition of system Xc-, have been confirmed to exhibit cancer effects. Ferroptosis strengthened the anticancer effect of the apoptosis induced by cisplatin in head and neck cancer cells, indicating that ferroptosis inducers could be used to enhance the effect of traditional anticancer drugs(1-3).
Conflict of interest: none declared
Financial support: none declared
This work is permanently accessible online free of charge and published under the CC-BY.

Bibliografie
-
Guo Q, Li L, Hou S, et al. The Role of Iron in Cancer Progression. Front Oncol. 2021;11:778492. doi:10.3389/fonc.2021.778492.
-
Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 2019;10(5):331. doi:10.1038/s41419-019-1564-7.
-
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266-282. doi:10.1038/s41580-020-00324-8
-
Bano I, Horky P, Abbas SQ, et al. Ferroptosis: A New Road towards Cancer Management. Molecules. 2022;27(7):2129. doi:10.3390/molecules27072129.
-
Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072. doi:10.1016/j.cell.2012.03.042.
-
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107-125. doi:10.1038/s41422-020-00441-1.
-
Vogt AS, Arsiwala T, Mohsen M, Vogel M, Manolova V, Bachmann MF. On Iron Metabolism and Its Regulation. Int J Mol Sci. 2021;22(9):4591. doi:10.3390/ijms22094591.
-
Jung M, Mertens C, Tomat E, Brüne B. Iron as a Central Player and Promising Target in Cancer Progression. Int J Mol Sci. 2019;20(2):273. doi:10.3390/ijms20020273.
-
Tifoun N, De Las Heras JM, Guillaume A, Bouleau S, Mignotte B, Le Floch N. Insights into the Roles of the Sideroflexins/SLC56 Family in Iron Homeostasis and Iron-Sulfur Biogenesis. Biomedicines. 2021;9(2):103. doi:10.3390/biomedicines9020103.
-
Filipovic MR, Koppenol WH. The Haber-Weiss reaction – The latest revival. Free Radic Biol Med. 2019;145:221-222. doi:10.1016/j.freeradbiomed.2019.09.017.
Articole din ediţiile anterioare
LETTER TO EDITOR | Ediţia 2 39 / 2017
The 3rd edition of General Oncology - to new biological and therapeutic dimensions of cancer
Alexandru Grigorescu
In recent years, progress and increased access to DNA sequencing technologies have opened a new era in molecular medicine and have revolutionized t...
17 mai 2017
VARIA | Ediţia 3 / 2015
Cancerul, o problemă nerezolvată
Doru Paul
Pornind de la ideea că eşecul nostru de a vindeca cancerul vine din lipsa noastră de a înţelege complexitatea sa, ştiinţa contemporană priveşte tre...
24 octombrie 2015
ORIGINAL STUDY | Ediţia 3 / 2016
Studiul erorilor diagnostice și terapeutice corelate cu studiul markerilor tumorali cu impact în prognosticul pacienților cu cancer localizat la cap și gât
Ioan Andrei, Anda Crişan, Florinel Bădulescu, Michael Schenker
Introducere. Alegerea protocolului de tratament pentru pacienții cu carcinoame ale sferei ORL stadii avansate locoregional, rămâne o discuție desc...
07 martie 2017
CASE PRESENTATION | Ediţia 1 58 / 2022
Intratumoral heterogeneity in breast cancer – a real challenge for diagnosis and treatment
Marius Pirciu, Elena Avram, Iolanda Georgiana Augustin, Daniela Zob, Andreea Lăzescu
Prezentăm cazul unei paciente cu neoplasm mamar la care, în evoluţia bolii, prin biopsii multiple, s-a remarcat o pronunţată heterogenitate in...
25 martie 2022