Pulmonary carcinoid tumors are rare low-grade malignant tumors which develop from the neuroendocrine system. These tumors represent 1% to 2% of all lung cancers and are classified as typical and atypical tumors. Approximately 80% of all pulmonary carcinoid tumors are low-grade typical carcinoids, and 20% are atypical carcinoid tumors. Most carcinoids arise from the central airways, are always malignant and generally have a slow growth and a high potential to metastasize. Only a minority of carcinoids are found in the lung periphery. We report the case of an 18-year-old female patient who presented with a decrease in the visual acuity of the right eye. After the ophthalmologic examination discovered a tumoral mass on the right eye, furthered examinations were performed. The chest radiography performed at the admission in the hospital revealed a tumoral mass situated in the right pulmonary hilum. A standard echography was also recommended which revealed multiple hepatic lesions. Afterwards, a CT scan was performed and confirmed the presence of the lesions from the right hilum and from the liver, as well as the invasive lesion located in the right ocular globe, and discovered a new pituitary tumor. The biopsy from the primary tumoral mass situated on the upper lobe of the right lung revealed the presence of a well-differentiated neuroendocrine tumor and the immunohistochemical tests were positive for SSTR2 and SSTR5, confirming the diagnosis of typical carcinoid. After being diagnosed, the patient started chemotherapy with cisplatin and etoposide, subsequently adding a somatostatin analogue and interferon, but under this therapy disease progression was observed, and the decision was to continue the second-line chemotherapy with capecitabine and temozolomide and long-acting analogue of somatostatin.
Neuroendocrine tumor (bronchial carcinoid) discovered after ocular symptoms – a case report
Tumoră neuroendocrină (carcinoid bronşic) descoperită în urma unor simptome oculare – prezentare de caz
First published: 13 decembrie 2022
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/OnHe.61.4.2022.7412
Abstract
Rezumat
Tumorile de tip carcinoid localizate la nivel pulmonar sunt tumori maligne rare, cu grad scăzut de diferenţiere, ce se dezvoltă de la nivelul sistemului neuroendocrin. Aceste tumori reprezintă 1% până la 2% din totalul cancerelor pulmonare şi sunt clasificate în tipice şi atipice. Aproximativ 80% din totalul tumorilor carcinoide pulmonare prezintă un grad scăzut de diferenţiere, reprezentând tumori carcinoide tipice, în timp ce restul de 20% sunt tumori carcinoide atipice. Majoritatea tumorilor carcinoide se dezvoltă la nivelul căilor respiratorii centrale, sunt întotdeauna maligne şi în general au creştere lentă şi potenţial de metastazare crescut, în timp ce doar o mică parte a tumorilor carcinoide pulmonare sunt localizate periferic. Prezentăm cazul unei paciente în vârstă de 18 ani care s-a prezentat acuzând scăderea acuităţii vizuale la nivelul ochiului drept. În urma consultului oftalmologic, s-a descoperit prezenţa unei formaţiuni tumorale situate la nivelul globului ocular drept, motiv pentru care alte investigaţii au fost efectuate. Radiografia pulmonară efectuată la internare a evidenţiat prezenţa unei formaţiuni tumorale situate în adiacenţa hilului pulmonar drept. O examinare ecografică a fost, de asemenea, recomandată, evidenţiind prezenţa unor multiple leziuni diseminate la nivel hepatic. Examenul CT efectuat ulterior a confirmat prezenţa formaţiunii tumorale hilare drepte şi a multiplelor leziuni hepatice, precum şi prezenţa formaţiunii tumorale localizate la nivelul globului ocular drept, dar a descoperit şi prezenţa unei formaţiuni hipofizare. Examenul biopsic al formaţiunii tumorale primare situate la nivelul pulmonar drept a evidenţiat prezenţa unei tumori neuroendocrine bine diferenţiate, iar testele imunohistochimice au fost pozitive pentru SSTR2 şi SSTR5, confirmând diagnosticul de tumoră carcinoidă tipică. După ce a fost diagnosticată, pacienta a început tratamentul chimioterapic cu cisplatin şi etopozidă, la care s-au adăugat analogi de somatostatină şi interferon, însă după confirmarea progresiei bolii, s-a luat decizia de a se începe tratamentul chimioterapic de linia a doua cu capecitabină şi temozolomid, alături de continuarea tratamentului cu analogi de somatostatină.
Introduction
Pulmonary carcinoid tumors are epithelial neuroendocrine tumors (NETs) that account for 1-2% of all lung cancers. They originate in the enterochromaffin cells (Kulchitsky cells) that are disseminated in the bronchopulmonary tract(1). They are classified into typical and atypical forms, and 80% of the cases are usually centrally located in the lungs. These types of tumors are always malignant, and generally present a slow growth and an increased rate of metastasis(2). Deletions on chromosome 11q are one of the few known genetic risk factors that promote their occurrence(3). The laboratory diagnosis is mainly based on three tests: 5-hydroxy-indol-acetic acid, chromogranin A, and the Ki67 marker, which classifies neuroendocrine tumors into three degrees of differentiation: NET G1 (Ki67<2%), NET G2 (Ki67=2-20%) and NET G3 (Ki67>20%)(4). There are numerous imaging methods that can provide the diagnosis of NETs, such as computed tomography (CT), magnetic resonance imaging (MRI), radiolabeled somatostatin analogue scintigraphy and SPECT/CT (111In-DTPA Pentetrotide, 99mTc-EDDA/HYNIC-TOC), and PET/CT with 68Ga-DOTA-peptides or 18F-FDG. When considering the radiotracers available for nuclear medicine investigations, 111In-DTPA Pentetrotide binds especially to subunits 2 and 5 of the somatostatin receptors, while 99mTc-EDDA/HYNIC-TOC has high affinity for subunit 2 of somatostatin receptors and low affinity for subunits 3 and 5(5-7). 68Ga-DOTA-peptides are composed of three major peptides: TOC, NOC, TATE; all three peptides have affinity for subunits 2 and 5, while NOC presents also affinity for subunit 3 of somatostatin receptors(8). Furthermore, PET/CT with 18F-FDG has an increased affinity for tumors with a higher grade of differentiation (NET G2-G3)(6). At present, SPECT-CT represents an alternative method of diagnosis, with similar sensitivity and specificity to 68Ga-PET-CT, making this procedure a good follow-up investigation for patients with neuroendocrine tumors. The surgical treatment may be recommended for treating the primary site and for liver metastases, regardless of tumoral grade(9,10). Other treatment options comprise chemotherapy, biological therapy, long- or short-acting analogs of somatostatin, chemoembolization (embolization with Yttrium-90 microspheres – especially for hepatic metastases), peptide receptor radionuclide therapy (PRRT) and radiotherapy(11-14).
Case presentation
We present the case of an 18-year-old female patient with a typical bronchial carcinoid diagnosed in 2020, which presented at the onset a decrease in the visual acuity of the right eye. The ophthalmologic examination found an amelanotic macular tumoral mass on the right eye, with retinal detachment associated. Afterwards, the ocular ultrasonography confirmed the hyperechogenic choroidal mass with retinal detachment and choroidal melanoma was suspected.
After the ophthalmologic examination, the patient presented to our hospital for further investigations. At the admission, the standard evaluation protocol – which includes laboratory tests, chest radiography and the abdominopelvic ultrasonography – was performed. The laboratory tests performed were normal. The chest radiography revealed a tumoral mass situated in the right pulmonary hilum, while the abdominal ultrasonography revealed multiple hepatic lesions. After these results, a CT scan was indicated.
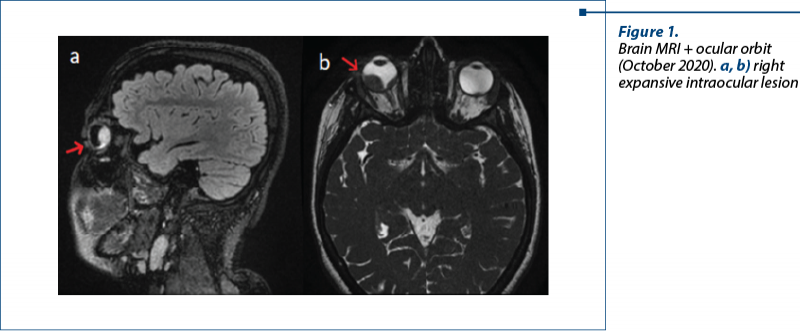
The CT scan (head, thorax, abdominal, pelvic) performed in October 2020 revealed a primary tumoral mass in the right pulmonary hilum, extended to the upper lobe, and other lesions in the liver, as well as invasive lesions located in the right ocular globe and hypophysis.
For the hypophyseal and ocular lesions found on the CT examination, a brain MRI was recommended and performed. This revealed on the right eye a trilobate formation with a crescent-shaped appearance, plated on the posterior choroid.
In October 2020, the patient performed a bronchoscopy which described complete obstruction of the upper lobe of the right lung by a tumoral process. The biopsy from the primary tumoral mass revealed the presence of a well-differentiated neuroendocrine tumor (Ki67=10%) and the immunohistochemical tests were positive for SSTR2 and SSTR5.
The histopathological and immunohistochemical tests confirmed the diagnosis: typical carcinoid.
The neuroendocrine whole-body scintigraphy and SPECT-CT performed in November 2020 highlighted the presence of somatostatin receptors in the tumoral mass situated in the right pulmonary hilum, in the hepatic lesions and in the ocular and pituitary metastases.
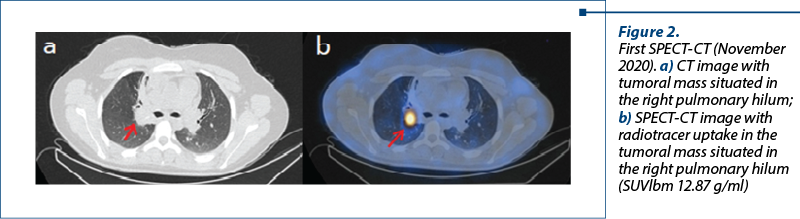
At the time of the first scintigraphy, the paraclinical data showed increased serotonin values – 456.55 µg/L (2590.78 nmol/L), and normal values of chromogranin A (63.03 ng/ml) and of 5-hydroxy-indol-acetic acid (7.02 mg/24 h; 36.71 µmol/24 h).
The patient underwent six series of chemotherapy with cisplatin and etoposide from December 2020 to April 2021, subsequently adding a somatostatin analogue and interferon (from January 2021). Also, she received additional palliative radiotherapy for the right eye, which resulted in a decrease in the tumoral size at this level.
The SPECT-CT performed after six months of treatment (in April 2021) showed no significant changes in the primary tumoral mass, but revealed a notably increased uptake in the ocular and pituitary metastases.
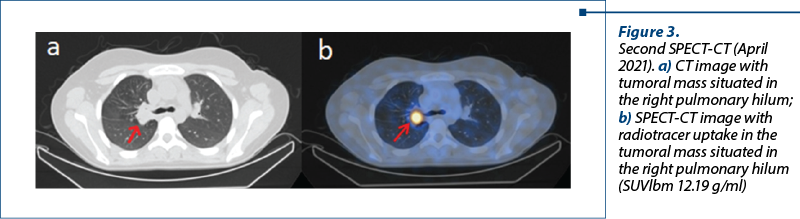
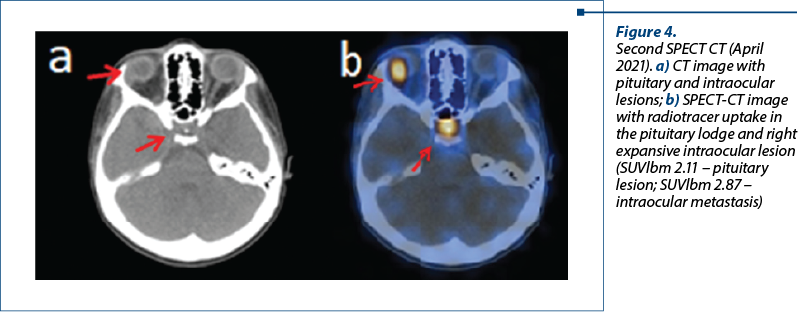
The laboratory tests performed in September 2021, before the third SPECT-CT and after six series of chemotherapy with cisplatin and etoposide and long-acting analogue of somatostatin treatment (January 2021 – September 2021), showed normal values of serotonin, 5-hydroxy-indol-acetic acid and of chromogranin A, but increased compared to June 2021.
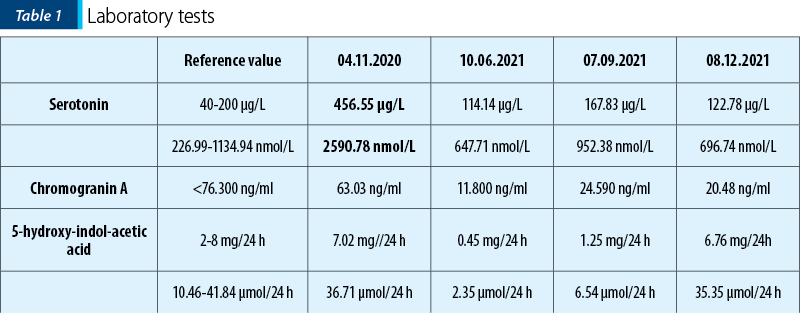
The third SPECT-CT examination, performed in October 2021, showed a decrease in radiotracer’s uptake in the ocular and hypophyseal metastases, but revealed two new lesions in hepatic segments IV and VIII, confirming disease progression.
![Figure 6. a) Third SPECT-CT (October 2021); b) Fourth SPECT-CT (April 2022). Reduction of the radiotracer uptake in the pituitary lodge and right expansive intraocular lesion (SUVlbm 5.42 g/ml versus 1.47 g/ml [a versus b] – pituitary lesion; SUVlbm 3.11 g/ml versus 1.34 g/ml [a versus b] – intraocular metastasis)](/image/24455/0/figure_6_24455.png)
![Figure 7. a) Third SPECT-CT (October 2021); b) Fourth SPECT-CT (April 2022). Significant reduction in radiotracer uptake in the right lung mass (SUVlbm 10.8 g/ml versus 3.60 g/ml [a versus b])](/image/24456/0/figure_7_24456.png)

After the third SPECT-CT examination that revealed the progression of disease, in November 2021 it was taken the decision to continue the chemotherapy treatment of second line (capecitabine and temozolomide), along with long-acting analogue of somatostatin.
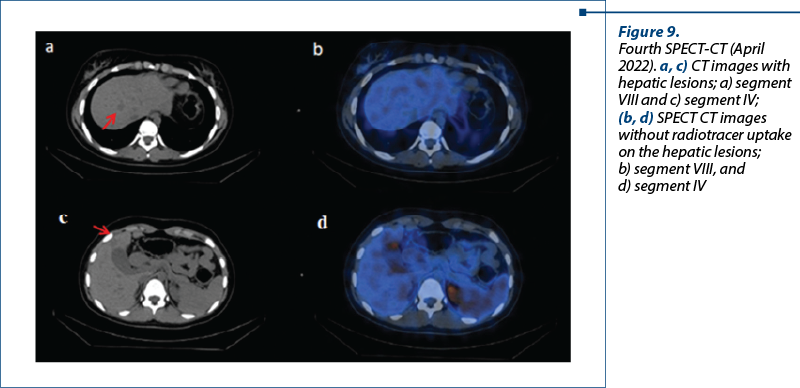
The fourth SPECT-CT examination from April 2022 revealed a significant reduction in radiotracer’s uptake in the right lung mass (currently SUVlbm 3.60 g/ml versus 10.8 g/ml in the previous exam), right intraocular lesion (currently SUVlbm 1.34 g/ml versus the previous one of 3.11 g/ml) and in the pituitary lesion (current SUVlbm 1.47 g/ml versus 5.42 g/ml previously), and also the disappearance of radiotracer uptake from the hepatic segments IV and VIII.
Discussion
Neuroendocrine tumors include malignancies with different origins, clinical presentations and prognosis. Ocular metastases from NETs are exceptionally rare, and originate more frequently from foregut primary tumors (lung, esophageal, and thymic NETs). The most common ocular site of secondary lesions is the uvea, likely due to its high vascularization. Uveal metastases usually involve the choroid (88% of cases) and infrequently the iris (9% of cases) or the ciliary body (2% of cases)(15). When neuroendocrine tumors metastasize to the ocular region, gastrointestinal NET typically spreads to the orbit, whereas bronchial NET classically metastasizes to the choroid(15).
Shields et al. described in their case report three cases of neuroendocrine tumors with ocular metastasis as first sign of disease. All three patients accused blurred vision and performed an ophthalmologic examination and ocular ultrasound which found an amelanotic choroidal mass. After further investigations, the diagnosis of bronchopulmonary neuroendocrine tumor with carcinoid intraocular metastasis was made in all three cases, after excluding the diagnosis of amelanotic melanoma(16).
In the case of our patient, the ophthalmologic exam found on the right eye a macular tumor formation with peritumoral retinal detachment and, after the ocular ultrasound, malignant choroidal melanoma was suspected.
Yiu et al. presented a case of choroidal metastasis from a neuroendocrine tumor masquerading as choroidal melanoma. They presented the case of a patient with a large choroidal metastasis from a pancreatic neuroendocrine tumor that showed a collar-button configuration which was classically suggestive for melanoma(17).
Ocular symptoms and signs may be initially present, further imaging and paraclinical examinations confirming the presence of metastases at this level and the primary neuroendocrine tumor. A broad differential and careful consideration of ocular and systemic symptoms are critical in such challenging cases. Shen et al. published a case report which contains two cases of metastatic neuroendocrine tumors masked as primary ocular disease(18). The first case presented a 38-year-old man who was referred with subacute onset diplopia and fluctuating ptosis suggestive of myasthenia gravis, and the second case was a 21-year-old man who presented with blurry vision and was found to have a pigmented ciliary body mass and retinal detachment suggestive of uveal melanoma. Both patients were ultimately diagnosed with metastatic neuroendocrine tumors(18).
The pituitary gland is another rare site where neuroendocrine tumors metastasize. Pituitary metastases are uncommon findings and are mainly derived from breast and lung cancers. No extensive reviews of pituitary metastases from neuroendocrine tumors have been recorded. Ragni et al. described a clinical case of pituitary metastases from pancreatic NET and reviewed the clinical features of pituitary metastases from NETs reported in the literature(19). He reported the case of a 59-year-old female patient, previously submitted to duodeno-cephalo-pancreatectomy for a well-differentiated pancreatic NET, with known hepatic metastases, who underwent a 68 Ga-DOTATOC PET/CT that revealed an uptake in the pituitary gland. A subsequent MRI revealed a pituitary lesion, with suprasellar extension. After a hormonal and genetic diagnostic workup that excluded the diagnosis of MEN 1, the worsening of headache and visual impairment and the growth of the lesion led to its surgical removal, and a pituitary localization of the pancreatic NET was identified(19).
Regarding the published cases of pituitary metastases from NETs, the most common tumor type was small cell lung cancer (SCLC), accounting for nearly half of the cases, followed by bronchial and pancreatic well differentiated NETs. The most frequent symptom was a variable degree of visual impairment, while headache was reported in half of the cases. Partial or total anterior hypopituitarism was present in approximately three quarters of the cases, while diabetes insipidus was less common. The most frequent treatment for pituitary metastases was surgical resection, followed by radiotherapy and chemotherapy. The clinical outcome was in line with previous reports of pituitary metastases from solid tumors, with a median survival of 14 months. Surgery of pituitary metastases was associated with prolonged survival. Pituitary metastases from NETs have clinical features similar to those of metastases derived from other solid tumors, although the involvement of the anterior pituitary seems more frequent; a thorough pituitary hormonal evaluation is mandatory, after focused radiological studies, particularly if a surgical approach is considered. The optimal management of pituitary metastases remains disputed and seems mainly driven by the aggressiveness of the primary tumor and the presence of symptoms. In well-differentiated NETs, particularly in the case of symptomatic pituitary metastases, surgical removal may be a reasonable approach(19).
The most frequent localization of neuroendocrine tumor metastases is the liver. Nearly half of all patients with extrahepatic primary cancer have hepatic metastases. The severe prognostic implications of hepatic metastases have made surgical resection an important first-line treatment in the NET management. However, limitations, such as the presence of extrahepatic metastases or the presence of liver failure, exclude the majority of patients as surgical candidates, leaving chemotherapy and locoregional therapies as next best options. Selective internal radiation therapy (SIRT) is a form of catheter-based locoregional cancer treatment for unresectable tumors, involving transarterial injection of microspheres embedded with 90Y. The therapeutic radiation dose is selectively delivered as the microspheres permanently embed themselves within the tumor vascular bed. The use of SIRT has been conventionally aimed at treating primary hepatic tumors (hepatocellular carcinoma) or colorectal and neuroendocrine metastases(20). Liver radioembolization with 90Y-microspheres (TARE) is reserved for nonoperable patients usually due to high tumor burden(21). Jia et al. performed a systematic review on 90Y-radioembolization for unresectable metastatic neuroendocrine liver tumor(22). A total of 11 studies and seven abstracts, involving 870 patients, were included in the final analysis. In 11 of these studies, 19.8% (77/388) of patients had undergone transarterial bland embolization (TABE) or transarterial chemoembolization (TACE) before 90Y therapy. The median disease control rate among all patients was 86% at three months after 90Y therapy. The median survival was 28 months, with 1-year, 2-year, and 3-year survival rates of 72.5%, 57% and 45%, respectively. The median survival values for patients who received resin- and glass-based 90Y treatment were 27.6 and 31.7 months, respectively. The survival values for patients with carcinoid, pancreatic and unclassified origin of NETs were 56, 31 and 28 months, respectively; the survival values for patients with grade I, II and III NETs were 71, 56 and 28 months, respectively. Carcinoid syndrome was reported in 52.4% (55/105) of patients, and 69.1% of those with clinical symptoms demonstrated improvement in symptoms after 90Y radioembolization. Complications were reported in nine studies, including radiation gastritis (n=4), duodenal ulcer (n=2), death due to liver failure (n=1), and radiation cholecystitis (n=1). The most common side effects were abdominal pain (median: 32.6%), nausea/vomiting (median: 32.5%), and fatigue (median: 30.4%). This treatment is also effective for patients who have undergone unsuccessful TABE/TACE therapy and for the relief of symptoms in patients with carcinoid syndrome(22).
Although the symptoms and signs are complex and of great variety, the majority of patients with pulmonary carcinoid have an excellent survival rate, even though they present lymph node metastases at diagnosis. Neuroendocrine tumors of the lung represent a broad spectrum of morphologic types that share specific morphologic, immunohistochemical, ultrastructural, and molecular characteristics. These tumors comprise approximately 20% of all primary lung cancers(23).
Conclusions
The case presented in our paper is particular, considering its initial symptoms at the onset, through visual impairment, and the localization of the metastases. Neither the ocular globe, nor the hypophysis represent common metastasizing sites for neuroendocrine tumors, thus making this case study one of reference when considering peculiar presentations of neuroendocrine tumors.
Out of over 300 cases examined in our clinic, this is the only one with such an atypical presentation, raising the issue of differential diagnosis. The evolution of the disease was stable in the beginning, but then it progressed, with no significant therapeutic response to the two lines of treatment used.
Conflicts of interests: The authors declare no conflict of interests.
Bibliografie
-
Caplin ME, Baudin E, Ferolla P, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26(8):1604-1620. doi:10.1093/annonc/mdv041.
-
Limaiem F, Tariq MA, Wallen JM. Lung Carcinoid Tumors. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2022.
-
Zuetenhorst JM, Taal BG. Metastatic Carcinoid Tumors: A Clinical Review. Oncologist. 2005;10:123–131, doi:10.1634/theoncologist.10-2-123.
-
Rindi G, Mete O, Uccella S, et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr Pathol. 2022;33(1):115-154. doi:10.1007/s12022-022-09708-2.
-
Mikołajczak R, Maecke HR. Radiopharmaceuticals for Somatostatin Receptor Imaging. Nuclear Medicine Review. 2016;19:126–132, doi:10.5603/NMR.2016.0024.
-
Gherghe M, Lazăr AM, Stanciu AE, et al. The New Radiolabeled Peptide 99mTcEDDA/HYNIC-TOC: Is It a Feasible Choice for Diagnosing Gastroenteropancreatic NETs?. Cancers (Basel). 2022;14(11):2725. doi:10.3390/cancers14112725.
-
Garai I, Barna S, Nagy G, Forgacs A. Limitations and pitfalls of 99mTc-EDDA/HYNIC-TOC (Tektrotyd) scintigraphy. Nucl Med Rev Cent East Eur. 2016;19(2):93-98. doi:10.5603/NMR.2016.0019.
-
Ambrosini V, Fanti S. 68Ga-DOTA-Peptides in the Diagnosis of NET. PET Clin. 2014;9:37–42. doi:10.1016/j.cpet.2013.08.007.
-
Watzka FM, Fottner C, Miederer M, et al. Surgical Treatment of NEN of Small Bowel: A Retrospective Analysis. World J Surg. 2016;40(3):749-758. doi:10.1007/s00268-016-3432-2.
-
Farley HA, Pommier RF. Surgical Treatment of Small Bowel Neuroendocrine Tumors. Hematol Oncol Clin North Am. 2016;30:49–61. doi:10.1016/j.hoc.2015.09.001.
-
Eriksson B, Annibale B, Bajetta E, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: chemotherapy in patients with neuroendocrine tumors. Neuroendocrinology. 2009;90(2):214-219. doi:10.1159/000225950.
-
Oberg K, Ferone D, Kaltsas G, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: biotherapy. Neuroendocrinology. 2009;90(2):209-213. doi:10.1159/000183751.
-
Hicks RJ, Kwekkeboom DJ, Krenning E, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasia: Peptide Receptor Radionuclide Therapy with Radiolabeled Somatostatin Analogues. Neuroendocrinology. 2017;105(3):295-309. doi:10.1159/000475526.
-
Lewandowski RJ, Toskich BB, Brown DB, El-Haddad G, Padia SA. Role of Radioembolization in Metastatic Neuroendocrine Tumors. Cardiovasc Intervent Radiol. 2022;45(11):1590-1598. doi:10.1007/s00270-022-03206-y.
-
Shields JA, Shields CL. Metastatic tumors to the uvea, retina, and optic disc.In: Shields JA, Shields CL. Intraocular Tumors: An Atlas and Textbook. Philadelphia, PA Lippincott Williams & Wilkins; 198-227
-
Shields CL, Say EA, Stanciu NA, Bianciotto C, Danzig CJ, Shields JA. Cavitary choroidal metastasis from lung neuroendocrine tumor: report of 3 cases. Arch Ophthalmol. 2011;129(1):102-4. doi: 10.1001/archophthalmol.2010.329.
-
Yiu G, Cummings TJ, Mruthyunjaya P. Choroidal metastatasis from a neuroendocrine tumor masquerading as choroidal melanoma. Ophthalmic Surg Lasers Imaging Retina. 2014;45(5):456-458. doi:10.3928/23258160-20140725-01.
-
Shen A, Haghighi A, Liang T, et al. Metastatic neuroendocrine tumors mimicking as primary ocular disease. Am J Ophthalmol Case Rep. 2022;26:101425. doi:10.1016/j.ajoc.2022.101425.
-
Ragni A, Nervo A, Papotti M, et al. Pituitary metastases from neuroendocrine neoplasms: case report and narrative review. Pituitary. 2021;24(5):828-837. doi:10.1007/s11102-021-01178-9.
-
Kuei A, Saab S, Cho SK, Kee ST, Lee EW. Effects of Yttrium-90 selective internal radiation therapy on non-conventional liver tumors. World J Gastroenterol. 2015;21(27):8271-8283. doi:10.3748/wjg.v21.i27.8271.
-
Tudela-Lerma M, Orcajo-Rincón J, Ramón-Botella E, et al. Efficacy and safety of Yttrium-90 radioembolization in the treatment of neuroendocrine liver metastases. Long-term monitoring and impact on survival. Eficacia y seguridad de la radioembolización con 90Y en el tratamiento de metástasis hepáticas de tumores neuroendocrinos. Seguimiento a largo plazo y repercusión en la supervivencia. Rev Esp Med Nucl Imagen Mol (Engl Ed). 2021;40(2):82-90. doi:10.1016/j.remn.2020.09.002.
-
Jia Z, Wang W. Yttrium-90 Radioembolization for Unresectable Metastatic Neuroendocrine Liver Tumor: A Systematic Review. Eur J Radiol. 2018;100:23–29, doi:10.1016/j.ejrad.2018.01.012.
-
Langfort R, Rudziński P, Burakowska B. Rozrosty neuroendokrynne płuc. Histologiczne spektrum podtypów, aktualne pglady dotyczace rozpoznawania i leczenia [Pulmonary neuroendocrine tumors. The spectrum of histologic subtypes and current concept on diagnosis and treatment]. Pneumonol Alergol Pol. 2010;78(1):33-46.
Articole din ediţiile anterioare
Variability of response to multimodal therapy in a stage IV bronchial carcinoid: a case report
Neoplasmele pulmonare neuroendocrine (NEN) se clasifică în tumori neuroendocrine (NET) şi carcinoame neuroendocrine (NEC). NET cuprind tumo...
Neuroendocrine tumors of the small bowel: a literature review
Cancerul neuroendocrin, adesea cunoscut sub numele de tumoră neuroendocrină (NET) sau neoplasm neuroendocrin, provine din celulele specializate...