Immune thrombocytopenia is an autoimmune disorder that involves the platelet destruction in the spleen. Often, patients do not respond to conventional therapy. The management of refractory or relapsed immune thrombocytopenia poses a challenge for hematologist. We report the case of a 43-year-old patient in whom conventional treatment was not successful but who responded to TPO agonist eltrombopag.
CASE PRESENTATION
Refractory immune thrombocytopenia – case presentation
Trombocitopenia imună refractară – prezentare de caz
Iuliana Iordan,
Andreea Neculcea,
Stejara Nicoleta Mihai,
Diana Emanuela Bonea,
Andreea Spînu,
Alina Mititelu,
Claudiu Popescu,
Raluca Truican,
Anca Nicolescu,
Ana Maria Prof. Dr. Vlădăreanu
First published: 31 mai 2022
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/OnHe.59.2.2022.6541
Abstract
Rezumat
Trombocitopenia imună este o boală autoimună care implică distrugerea trombocitelor în splină. Adesea, pacienţii nu răspund la terapia convenţională. Managementul trombocitopeniei imune refratare sau recăzute reprezintă o provocare pentru hematolog. Prezentăm cazul unui pacient în vârstă de 43 de ani, cu răspuns nesatisfăcător la terapia convenţională, dar care a răspuns la agonistul TPO eltrombopag.
Introduction
Immune thrombocytopenia (ITP) is an autoimmune disorder characterized by an immune-based thrombocytopenia. The mechanisms involve both increased immune-mediated destruction in the spleen and reduced production of platelets in the bone marrow. The etiology of thrombocytopenia often involves autoimmune pathologies, viral infections, Helicobacter pylori and lymphoproliferative neoplasms, but in most cases there is an idiopathic ITP(1).
The current classification of ITP, depending on the time since diagnosis, defines three types of ITP: newly diagnosed – up to three months since diagnosis, persistent – three to 12 months since diagnosis, and chronic ITP – more than 12 months since diagnosis(1).
The current treatment of ITP is step-based, strictly regimented in ITP, according to the 2019 ASH guidelines. First-line therapy in ITP includes corticosteroids, intravenous immunoglobulins and anti-Rh(D) antibodies. The goal of treatment is the reduction of bleeding complications and an increase of the platelet level of more than 20-30x109/L, with low drug toxicity(1).
Refractory ITP is challenging. It is often difficult to treat, particularly in patients with severe thrombocytopenia or active bleeding. Patients with refractory ITP respond poorly to a variety of treatments and may develop worsening disease and medication-induced toxicities. They may have markedly reduced quality of life and associate a high hemorrhagic and infectious morbidity and mortality.
Other treatment options, such as monoclonal antibody rituximab, immunosuppressive agents (e.g., azathioprine, cyclosporine), fostamatinib (splenic tyrosine kinase inhibitor), newer TPO agents and rapamycin, may be useful alone or in combination therapies. In some cases, the thrombopoietin-mimetic agents offer new perspectives in the treatment of refractory ITP, with higher response rates and lower toxicity(1-3).
Case presentation
We report the case of a 43-year-old male admitted to our hospital for fatigability, faintness and cutaneous hemorrhagic lesions. One month before admission, he was diagnosed with immune thrombocytopenia; he received a five-day pulse of corticosteroids, but with a modest response. The patient had no prior medical history or bleeding events and he did not take any other medication before admission. The clinical exam confirmed the hemorrhagic lesions, namely ecchymoses and purpura, and gum bleeding. No other clinical abnormalities were identified.
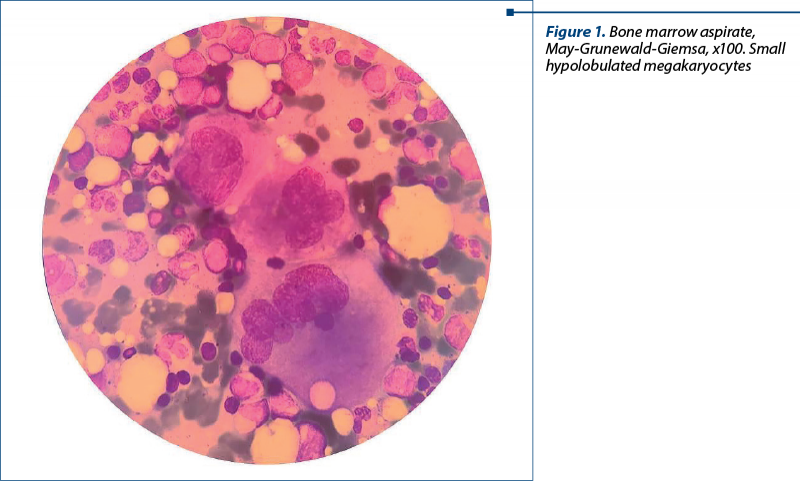
The complete blood count confirmed the presence of severe thrombocytopenia and leukocytosis (Table 1). We considered leukocytosis to be secondary to corticosteroid therapy.
The bone marrow aspirate was conclusive of a stimulated megakaryocytic lineage, with increased number of small megakaryocytes (Figure 1), and revealed no signs of dysplasia.
The computed tomography did not show any abnormalities.
We confirmed the previous diagnosis of primary immune thrombocytopenia. We repeated the pulse therapy with dexamethasone 24 mg/day for five days, but with an extremely modest response.
After two weeks, we administered intravenous immunoglobulin. The platelet count rose to normal up to 270 x109/L, but the increase was short, for only six days.
We switched the corticosteroid on metilprednisolone 32 mg/day and two doses of romiplostim 0.2 ml. The platelet number increased to 50x109/L and the patient was discharged. A week after, the patient came to the emergency room with severe thrombocytopenia (13x109/L) developed during treatment, and he was admitted to our clinic. We started pulse therapy with dexamethasone 24 mg/day for five days, but without significant changes in platelets number.
Considering the lack of response to corticotherapy, the short response to immunoglobulin and immunosuppressive therapy and the absence of other available pharmacological treatment options in that moment, we proposed splenectomy, after a discussion about the risks and benefits of this procedure. The patient was vaccinated with the 13-valent pneumococcal conjugate vaccine, the Hemophilus influenzae type b vaccine, the quadrivalent meningococcal conjugate ACWY vaccine, and the monovalent meningococcal serogroup B vaccine. Before splenectomy, an increased platelet level (50x109/L) was obtained using intravenous immunoglobulins and platelet transfusions. The surgical procedure was performed without any complications. After splenectomy, the platelets increased but staidly decreased at 25-30 x109/L, and the cutaneous hemorrhagic syndrome was still persistent. We decided to continue corticosteroids in low dosage, but without a therapeutic response after four weeks, the severe thrombocytopenia being persistent.
We considered the patient to be refractory to treatment. The “refractory” definition described patients whose platelet counts do not respond to two treatment lines, the platelet counts are very low and accompanied by bleeding.
Therefore, we decided to start the treatment with the TPO agonist eltrombopag. The clinical and biological response was favorable. The patient did not have any signs of bleeding and had a PLT of approximately 40-60 x109/L after two weeks of eltrombopag 25 mg/day.
Discussion
The most frequently used first-line therapy in ITP relies on corticosteroids. For most patients (60-80%), a response is obtained in 2-14 days(4,5). In our case, the patient did not respond to corticotherapy (the PLT remained about 5x109/L) and a duration of corticosteroid therapy over six weeks is not desirable due to the adverse events.
Another option for first-line treatment is intravenous immunoglobulins. The literature reports a response in 90% of patients, but with transient responses(4). After the failure with corticotherapy, we started intravenous immunoglobulins. The patient had a good response in a few days but it was not a persistent one; the response lasted about a week.
In Romania, rituximab is not approved for ITP second-line treatment. Thrombopoietin mimetics were approved for second-line and refractory ITP treatment, but can be used one year after the diagnosis of ITP.
Splenectomy is still used in the refractory ITP due to higher rates of durable responses, namely 60-70%(4). Despite the risk of complications, including procedure-related complications, infections, thromboembolism and malignancy, splenectomy is still considered a possible option for second-line treatment. The patient had a high risk for bleeding, with a persistent PLT level of about 5x109/L and a low-quality life. He was refractory to all treatments we used, except for intravenous immunoglobulins to which he had a transient response. The advantages and disadvantages were explained to the patient.
After splenectomy, a poor therapeutic response was obtained. Therefore, there was a need for a new therapeutic line.
Recently, the indications for eltrombopag moved from refractory ITP also to persistent ITP. It significantly reduces exposure to corticosteroid therapy and there is a real benefit for patients regarding also the quality of life. Eltrombopag is an oral thrombopoietin receptor agonist, proven to increase PLT in both adults and children with refractory ITP. Most cases require long-term usage of this drug, but in 30% of cases it can be discontinued without the loss of response(4).
The patient was treated with TPO agonist eltrombopag, with a favorable response. The PLT increased to a safer range of 40-60 x109/L, thus lowering the risk for hemorrhage. Eltrombopag was continued, the target of treatment was not the normalization of the platelets, but the reduction of the hemorrhagic syndrome.
Conclusions
Refractory ITP is a challenge for clinicians regarding treatment and monitoring, due to a significantly increased risk of drug-related toxicity and bleedings complications, including life-threatening events. With recent developments, especially thrombopoietin mimetics, an acceptable PLT can be achieved, with good tolerability and a low rate of adverse reactions. Combination strategies may target multiple pathogenetic mechanisms and trigger additive or synergistic effects and they can optimize the management of refractory ITP. However, not all patients respond to these therapies, and novel drugs are needed for this challenging pathology.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Onisâi M, Vlădăreanu AM, Spînu A, et al. Idiopathic thrombocytopenic purpura (ITP) – new era for an old disease. Rom J Intern Med. 2019;57(4):273-283.
-
Cuker A, Neunert CE. How I treat refractory immune thrombocytopenia. Blood. 2016;128(12):1547-1554.
-
Gonzalez-Porras JR, Bastida JM. Eltrombopag in immune thrombocytopenia: efficacy review and update on drug safety. Ther Adv Drug Saf. 2018;9(6):263–285.
-
Gomez-Almaguer D. Eltrombopag-based combination treatment for immune thrombocytopenia. Ther Adv Hematol. 2018;9(10):309–317.
-
Terrel DR, Neunert CE, Cooper N, et al. Immune Thrombocytopenia (ITP): Current Limitations in Patient Management. Medicina (Kaunas). 2020;56(12):667.
Articole din ediţiile anterioare
REVIEW | Ediţia 1 62 / 2023
Immune-mediated complications of monoclonal antibodies used in onco-hematology
Iuliana Iordan, Dan-Corneliu Jinga, Claudiu Dragoş Popescu, Ana Maria Vlădăreanu
Anticorpii monoclonali sunt tratamente cu eficienţă superioară pentru multiple afecţiuni hematologice şi oncologice. Chiar dacă majoritatea acestor...
23 martie 2023
SUPPORTIVE AND PALLATIVE CARE IN CANCER | Ediţia 4 49 / 2019
Corticoterapia – o metodă terapeutică importantă în terapia oncologică
Mihaela Dănciulescu, Cristian Lungulescu
Îmbunătăţirea calităţii vieţii pacienţilor oncologici, atât la cei diagnosticaţi în stadiile incipiente ale bolii, cât şi la cei aflaţi în stadiul ...
14 decembrie 2019
HEMATOLOGY | Ediţia 3 64 / 2023
Current management of relapsed/refractory immune thrombocytopenia
Alina Mititelu, Șef de lucrări dr. Minodora Onisâi, Anca Nicolescu, Ioachim Preda-Naumescu, Ana Maria Vlădăreanu
Trombocitopenia imună (TCI) este o afecţiune autoimună dobândită, cu potenţial hemoragic, caracterizată prin prezenţa autoanticorpilor antitrom...
26 octombrie 2023
LETTER TO THE EDITOR | Ediţia 1 54 / 2021
Patologie rară cu debut atipic şi abordare multidisciplinară
Tribus Laura Carina, Prof. Dr. Carmen Georgeta Fierbinţeanu-Braticevici, Andreea Marin, Ovidiu Calapod, Ana Maria Prof. Dr. Vlădăreanu, Conf. Dr. Horia Bumbea, Iuliana Iordan, Șef de lucrări dr. Minodora Onisâi
Limfohistiocitoza hemofagocitară (HLH) este un sindrom rar, ameninţător de viaţă, cauzat de o inflamaţie necontrolată şi de distrucţia ţesuturilor....
24 martie 2021