Myelofibrosis is a chronic BCR-ABL1-negative myeloproliferative neoplasm characterized by bone marrow fibrosis, inefficient hematopoiesis and extramedullary hematopoiesis. Since their approval in the treatment of myelofibrosis, JAK inhibitors had a positive impact, being the most effective treatment for improving symptomatology and spleen reduction. However, one should weight the benefits and the risks of this treatment, which include hematological toxicity, opportunistic infections, reactivation of latent infections, and an increased risk for chronic lymphoproliferative disorders. We report two cases of myelofibrosis patients treated with a JAK2 inhibitor who developed non-Hodgkin lymphoma and tuberculosis, respectively.
Special evolution in myelofibrosis – case reports
Evoluţie specială în mielofibroză – prezentări de cazuri
First published: 13 decembrie 2022
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/OnHe.61.4.2022.7411
Abstract
Rezumat
Mielofibroza este un neoplasm mieloproliferativ cronic BCR-ABL1-negativ, caracterizat prin fibroza măduvei osoase, hematopoieză ineficientă şi hematopoieză extramedulară. De la aprobarea lor în tratamentul mielofibrozei, inhibitorii JAK au avut un impact pozitiv, fiind cel mai eficient tratament pentru îmbunătăţirea simptomatologiei şi reducerea dimensiunilor splinei. Cu toate acestea, ar trebui să fie puse în balanţă beneficiile şi riscurile acestui tratament, care includ toxicitatea hematologică, infecţiile oportuniste, reactivarea infecţiilor latente şi riscul crescut de boli limfoproliferative cronice. Raportăm două cazuri ale unor pacienţi cu mielofibroză trataţi cu un inhibitor JAK2 care au dezvoltat limfom non-Hodgkin şi, respectiv, tuberculoză.
Introduction
Myelofibrosis (MF), which includes primary MF and post-essential thrombocythemia of polycythemia vera MF, is a BCR-ABL1-negative chronic myeloproliferative neoplasm characterized by bone marrow fibrosis, inefficient hematopoiesis and extramedullary hematopoiesis(1). Clonal populations produce cytokines and growth factors that contribute to bone marrow fibrosis and stromal abnormalities.
The identification of driver mutations in myelofibrosis is an essential step toward understanding the disease’s pathophysiology, the clinical heterogeneity and the therapeutic options. There are three oncogenic driver mutations in myelofibrosis, namely Janus kinase 2 (JAK2), calreticulin (CALR), and the myeloproliferative leukemia virus oncogene (MPL)(2).
JAK inhibitors changed dramatically the treatment, providing numerous benefits such as lowering splenomegaly, alleviating symptoms, and improving the quality of life(3). They cannot, however, change the course of the disease to acute leukemia(3). Moreover, the efficiency of JAK inhibitors has to be balanced with the increased risk of opportunistic infections and the reactivation of latent ones(4).
During the COVID-19 pandemic, ruxolitinib was revealed to have a beneficial effect. Ruxolitinib reduces inflammatory markers, the time spent on mechanical ventilation, the hospitalization length, and the requirement for vasopressor support(5). Patients with MF and COVID-19 infection should therefore continue to take ruxolitinib.
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the only viable curative treatment option for myelofibrosis. However, it has restricted indications(6).
We report two cases of myelofibrosis with a particular history, one of which, after 12 years of evolution, developed non-Hodgkin lymphoma, and another one that acquired an infectious complication, namely tuberculosis.
Case 1
We report the case of a 46-year-old patient diagnosed with CALR-positive primary myelofibrosis. His medical history included ankylosing spondylitis, which had been treated with gold salts 30 years before the hematological diagnosis. He had marked thrombocytosis at the time of diagnosis, as shown in Table 1. Following the confirmation of the diagnosis, cytoreductive therapy, with hydroxyurea and thromboreductin, was started.
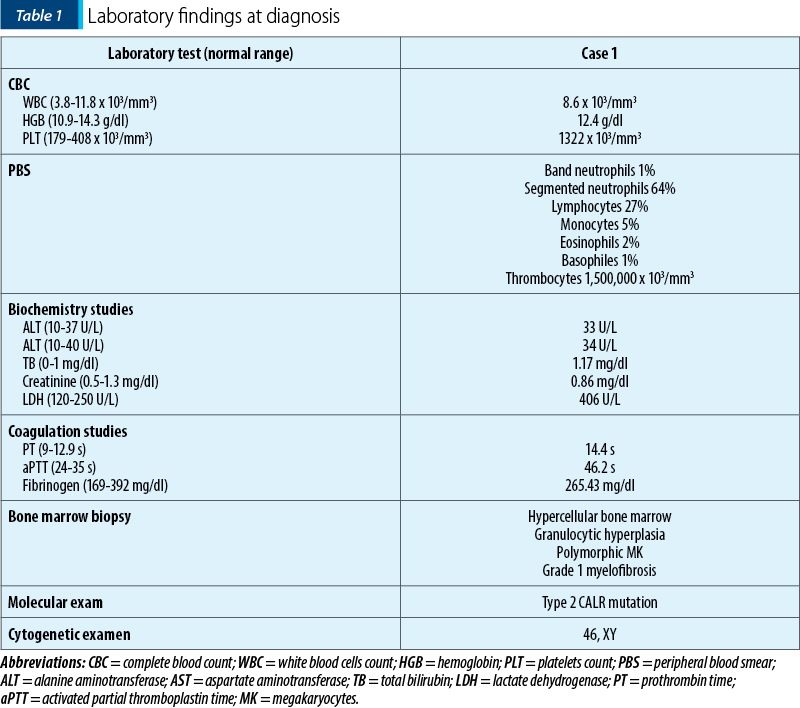
The patient decided to discontinue the treatment after three years. For two years, the patient did not take any myelofibrosis medication, and he also missed his scheduled check-ups. The patient developed hepato- and splenomegaly. At that time, a JAK2-inhibitor, ruxolitinib, was approved for symptomatic and high-risk myelofibrosis patients. The patient met the requirements and he started the drug.
After six years of ruxolitinib treatment, the patient came at the scheduled follow-up with fatigue, intolerance to physical activity and pallor. The complete blood count revealed moderate anemia (hemoglobin 8 g/dl). A bone marrow biopsy was performed. The results of the histological and immunohistochemical exams showed that the bone marrow was infiltrated by 55-60% marginal zone B-cells and also that myelofibrosis progressed to grade 3. Then, a computed tomography (CT) scan was performed to assess the disease’s extent. No adenopathies of other signs of organ infiltration were discovered, except for mild splenomegaly. Thus, we established the diagnosis of stage IV marginal-zone non-Hodgkin lymphoma. Taking into account the clinical and laboratory findings, we started rituximab therapy, in total six cycles, while maintaining ruxolitibib.
Discussion
Concurrent myeloid and lymphoid diseases might occur, which complicate the diagnosis and treatment. The most frequent association is between non-Hodgkin lymphoma and chronic myeloproliferative neoplasms (cMPN). In 52% of instances, the myeloproliferative disease comes first, followed by the lymphoproliferative disease(7). At ten years, the probability of developing chronic lymphoproliferation in MF patients is 2.96%, being 3.44-fold greater than in the general population(8). In our example, the patient was diagnosed with non-Hodgkin lymphoma 12 years after being diagnosed with primary myelofibrosis.
There are multiple possible hypotheses regarding the pathogenesis of lymphoproliferative diseases in cMPN patients. The first theory suggests that both diseases develop from the same lymphoid-myeloid progenitor. This hypothesis is supported by JAK2 V617F-positive patients’ increased likelihood of developing chronic lymphoproliferation. Furthermore, JAK2/STAT signaling is important in lymphomagenesis even when the genes are wild-type(9). Another hypothesis suggests that the two diseases are unrelated.
JAK1/2 inhibitors appear to increase the likelihood of developing aggressive B-cell lymphomas. JAK1/2 inhibition decreases the quantity and functionality of regulatory T cells, which results in immunosuppressive qualities that can aid in the development of lymphomas. Studies showed the presence of clonal B-cell population before beginning treatment. Therefore, recognizing the B-cell clone by flowcytometry, IgHV rearrangements investigation and PCR might be useful in identifying the at-risk individuals(10).
It is challenging to pinpoint which of the two disorders causes the symptoms, such as anemia, splenomegaly and catabolic signs. It might be difficult to decide which diseases to treat, but generally speaking, the condition with the worst symptoms should be addressed. Chemotherapy for chronic lymphoproliferations, however, can exacerbate cMPN patients’ anemia and decrease bone marrow reserve(11). The main symptoms of disease progression in our patient were anemia aggravation and splenomegaly. Given the findings of the bone marrow study, which showed worsening myelofibrosis and lymphoma infiltration, it is plausible to conclude that both illnesses contributed to the hematological picture. As a result, we chose to treat both disorders, and the patient’s condition improved.
Case 2
We report the case of a 66-year-old female patient who was initially diagnosed with JAK2 V617F essential thrombocythemia. At the time of the diagnostic, the patient had extensive thrombosis of the spleen, portal and mesenteric veins, as well as spleen infarctions. The laboratory tests at that moment are shown in Table 2.
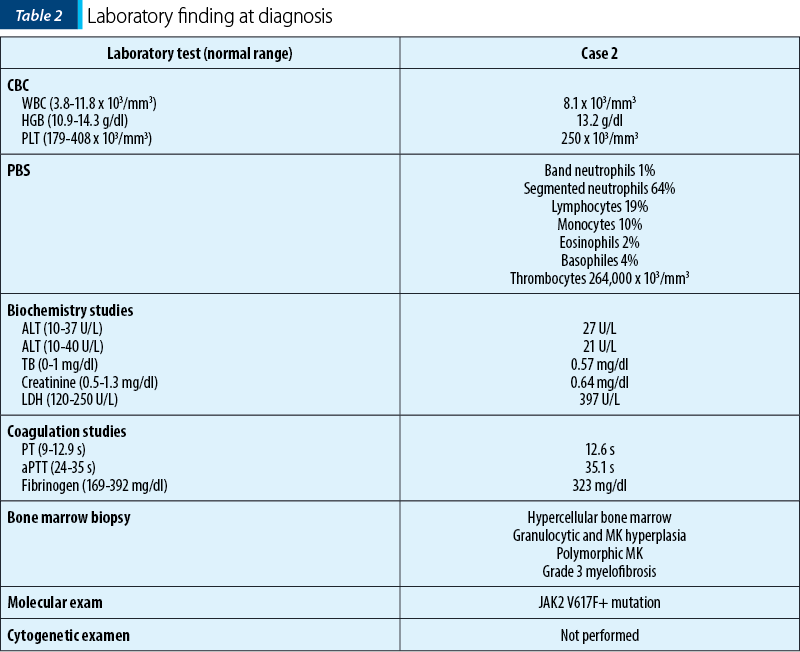
Three years later, she began to experience constitutional symptoms and developed mild anemia. We suspected the progression of the disease; therefore, we performed a bone marrow biopsy. We established the diagnosis of secondary myelofibrosis post-essential thrombocythemia based on the result of histopathological and immunohistochemical exam. The treatment with a JAK2-inhibitor – ruxolitinib – was initiated.
After one year, she described the aggravation of constitutional symptoms. Bilateral supraclavicular adenopathies were found during the clinical assessment. The adenopathies were firm and fixed in the deep tissue, with a maximum diameter of 2 cm. Laboratory tests showed worsening anemia and an elevated LDH (three times the upper limit of normal). Computed tomography was used to determine the extent of the adenopathies. Several profound adenopathies were discovered in the mediastinum and at the level of the abdomen, as well as a suspicious nodular lesion at the level of the left breast and a uterine mass (12.5/14 cm). For an accurate diagnosis, we performed a supraclavicular adenopathy biopsy. The histopathologic exam revealed the presence of numerous lesions with gigantic epithelial cells, some of which had caseous necrosis. The Ziehl-Neelsen stain identified acid-alcohol-resistant bacilli. We established the diagnosis of tuberculosis-related necrotizing granulomatous lymphadenitis with active lesions. The patient was started on antituberculosis medication. We used the lowest effective dose of ruxolitinib.
Discussion
Patients with myelofibrosis who are treated with ruxolitinib have an increased risk for infectious events, more often herpes zoster, urinary tract infections, pneumonia, sepsis and tuberculosis(4). JAK2 inhibition has numerous immunosuppressive mechanisms of immunosuppression, including reduced type 1 helper T cells response, impaired dendritic cell function, impaired natural killer cell maturation, and down regulation of regulatory T cells(12).
The infections with Mycobacterium tuberculosis in MF patients require a personalized treatment plan. Each case must be carefully analyzed in order to determine the appropriate treatment. If ruxolitinib cannot be stopped, it may be maintained in combination with antituberculosis medication at the lowest effective dosage. If we decide to discontinue ruxolitinib, we have to taper the doses to avoid discontinuation syndrome(12).
The patient in the second case had constitutional symptoms and multiple adenopathies. We considered a number of potential etiologies, including metastasis from another neoplasm, nodular clusters of myelofibrosis, myelofibrosis blast phase with nodular location, and coexistence of lymphoma. Even though tuberculosis-related lymphadenitis was not in our list of most probable diagnoses, it is important to consider this diagnosis in an immunosuppressed patient.
Mycobacterium tuberculosis can lead to a potentially fatal opportunistic infection in immunosuppressed patients. Therefore, it is extremely important to screen for a latent tuberculosis infection before beginning the treatment with a JAK2 inhibitor.
Conclusions
In myelofibrosis patients, the clinician must be aware of the potential of developing a chronic lymphoproliferative disease or an opportunistic infection. When new symptoms or clinical anomalies emerge in MF patients, we recommend further research. Patients with myelofibrosis who also have another condition should have their treatment personalized.
Conflicts of interests: The authors declare no conflict of interests.
Bibliografie
-
Mughal TI, Vaddi K, Sarlis NJ, Verstovsek S. Myelofibrosis-associated complications: pathogenesis, clinical manifestations, and effects on outcomes. Int J Gen Med. 2014 Jan 29;7:89-101. doi: 10.2147/IJGM.S51800.
-
Szuber N, Tefferi A. Driver mutations in primary myelofibrosis and their implications. Curr Opin Hematol. 2018 Mar;25(2):129-135. doi: 10.1097/MOH.0000000000000406.
-
Harrison CN, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Gisslinger H, Knoops L, Cervantes F, Jones MM, Sun K, McQuitty M, Stalbovskaya V, Gopalakrishna P, Barbui T. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016 Aug;30(8):1701-7. doi: 10.1038/leu.2016.148. Epub 2016 May 23. Erratum in: Leukemia. 2017 Mar;31(3):775.
-
Elli EM, Baratè C, Mendicino F, Palandri F, Palumbo GA. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front Oncol. 2019 Nov 7;9:1186. doi: 10.3389/fonc.2019.01186.
-
Quiros JR, Ross-Comptis J, Hathaway D 3rd, Sarfraz A, Sarfraz Z, Grigoryan Z, Romero KA, Gapizov A, Príncipe-Meneses FS, Somagutta MR, Riva-Moscoso A, Kapasi A. Ruxolitinib and the Mitigation of Severe COVID-19: A Systematic Review and Meta-analysis. Infect Chemother. 2021 Sep;53(3):436-448. doi: 10.3947/ic.2020.0126.
-
Tiribelli M, Palandri F, Sant’Antonio E, Breccia M, Bonifacio M. The role of allogeneic stem-cell transplant in myelofibrosis in the era of JAK inhibitors: a case-based review. Bone Marrow Transplant. 2020 Apr;55(4):708-716. doi: 10.1038/s41409-019-0683-1.
-
Bucelli C, Fattizzo B, Cattaneo D, Giannotta JA, Barbullushi K, Pasquale R, Barozzi E, Barbanti MC, Pettine L, Rossi FG, Reda G, Cassin R, Barcellini W, Baldini L, Iurlo A. Co-Occurrence of Myeloid and Lymphoid Neoplasms: Clinical Characterization and Impact on Outcome. A Single-Center Cohort Study. Front Oncol. 2021 Oct 18;11:701604. doi: 10.3389/fonc.2021.701604.
-
Vannucchi AM, Masala G, Antonioli E, Chiara Susini M, Guglielmelli P, Pieri L, Maggi L, Caini S, Palli D, Bogani C, Ponziani V, Pancrazzi A, Annunziato F, Bosi A. Increased risk of lymphoid neoplasms in patients with Philadelphia chromosome-negative myeloproliferative neoplasms. Cancer Epidemiol Biomarkers Prev. 2009 Jul;18(7):2068-73. doi: 10.1158/1055-9965.EPI-09-0353.
-
Rumi E, Baratè C, Benevolo G, Maffioli M, Ricco A, Sant’Antonio E. Myeloproliferative and lymphoproliferative disorders: State of the art. Hematol Oncol. 2020 Apr;38(2):121-128. doi: 10.1002/hon.2701.
-
Porpaczy E, Tripolt S, Hoelbl-Kovacic A, Gisslinger B, Bago-Horvath Z, Casanova-Hevia E, Clappier E, Decker T, Fajmann S, Fux DA, Greiner G, Gueltekin S, Heller G, Herkner H, Hoermann G, Kiladjian JJ, Kolbe T, Kornauth C, Krauth MT, Kralovics R, Muellauer L, Mueller M, Prchal-Murphy M, Putz EM, Raffoux E, Schiefer AI, Schmetterer K, Schneckenleithner C, Simonitsch-Klupp I, Skrabs C, Sperr WR, Staber PB, Strobl B, Valent P, Jaeger U, Gisslinger H, Sexl V. Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy. Blood. 2018 Aug 16;132(7):694-706. doi: 10.1182/blood-2017-10-810739. Epub 2018 Jun 14. Erratum in: Blood. 2019 Feb 14;133(7):768.
-
Bouchla A, Thomopoulos T, Papageorgiou S, Tsirigotis P, Bazani E, Gkirkas K, Vasilatou D, Glezou E, Stavroulaki G, Gkontopoulos K, Dimitriadis G, Pappa V. Coexistence of Myeloid and Lymphoid Neoplasms: A Single-Center Experience. Adv Hematol. 2019 Nov 3;2019:1486476. doi: 10.1155/2019/1486476.
-
Lescuyer S, Ledoux MP, Gravier S, Natarajan-Amé S, Duval C, Maloisel F, Mauvieux L, Toussaint E, Fornecker LM, Herbrecht R. Tuberculosis and atypical mycobacterial infections in ruxolitinib-treated patients with primary or secondary myelofibrosis or polycythemia vera. Int J Infect Dis. 2019 Mar;80:134-136. doi: 10.1016/j.ijid.2019.01.002.