Familial hypercholesterolaemia is the most common congenital metabolic disorder in children and adolescents. Although it is often underdiagnosed, the long-term consequences can be serious. Early diagnosis is therefore absolutely necessary and nutritional and therapeutic measures should be instituted immediately. Diagnostic criteria together with suggestive clinical signs are helpful to the clinician in diagnosing this group of pathologies.
REVIEW
Familial hypercholesterolemia in children and adolescents
Hipercolesterolemia familială la copii şi adolescenţi
First published: 30 iunie 2022
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Pedi.66.2.2022.6617
Abstract
Rezumat
Hipercolesterolemia familială reprezintă cea mai frecventă afecţiune metabolică congenitală la copii şi adolescenţi. Deşi de multe ori este subdiagnosticată, consecinţele pe termen lung pot fi grave. De aceea, diagnosticul precoce este absolut necesar, iar măsurile nutriţionale şi terapeutice trebuie instituite imediat. Criteriile de diagnostic împreună cu semnele clinice sugestive sunt de ajutor clinicianului în diagnosticarea acestui grup de patologii.
Familial hypercholesterolemia (FH) is an autosomal dominantly transmitted genetic disorder characterized by increased plasma levels of total cholesterol and LDL-cholesterol (LDLc) due to the reduction of the hepatic capacity to take over the LDL-cholesterol particles from the circulation(1).
There are two forms of the disease: heterozygous (HeFH) and homozygous (HoFH). The homozygous form is rare, being associated with clinical signs and with coronary heart disease during childhood. The heterozygous form is asymptomatic until adulthood when those affected develop events like premature cardiovascular disease(2,3).
FH pathophysiology
Familial hypercholesterolemia is associated with a mutation in one of three genes:
-
LDL receptor gene
-
apolipoprotein B gene
-
protein convertase subtilisin/kexin 9 gene (PCSK9).
The PCSK9 protein, synthesized mainly in the liver, has a role in regulating LDL hepatic receptors. After “loading” LDLc, they are internalized in the hepatocyte cytoplasm, where it releases LDLc to be metabolized, after which the receptor is recycled to the surface hepatocyte. If the receptor is bound to PCSK9, it is degraded.
Many recent studies present the ApoE and Stap1 genes as pathogenic variants for extremely rare forms of FH(4,5).
Epidemiology
FH is the most prevalent congenital metabolic disorder. The prevalence of familial hypercholesterolemia is reported as 1 in 500 individuals. The heterozygous phenotype is more common, with a prevalence of 1 in 200 individuals. Approximately 85-90% of patients with FH present mutations in the LDL receptor gene and about 15% of patients have mutations in the apolipoprotein gene. Approximately 5% of patients with FH have mutations in the PCSK9 gene(6).
Clinical manifestations of FH in children
The heterozygous form of familial hypercholesterolemia is often asymptomatic in the young population. They register lifetime increases in total cholesterol levels from 250 mg/dl to 500 mg/dl, and at the clinical examination of patients aged 20-39 years old with this form of the disease there were found: xanthomas in the Achilles tendon and in the tendons of the extensor muscles of the hand, senile arch corneal (gerontoxon), xanthelasma, clinical manifestations of coronary heart disease (after the age of 40 years old).
Although xanthomas are pathognomonic for familial hypercholesterolemia, they are not always present(7).
In the homozygous form of HF characterized by lack of LDL receptor activity, total cholesterol has elevated values, above 700 mg/dl, and this high level of plasma cholesterol can be met even in the newborn. Other features of this form are:
-
Xanthomas can exist from the first month of life.
-
Signs and symptoms of coronary heart disease appear in the first year of life.
-
Diffuse atherosclerosis in the peripheral arteries.
-
Aortic stenosis.
-
Cerebrovascular diseases.
Cardiovascular damage begins shortly after birth in children with familial hypercholesterolemia, through morphological and functional changes of the arterial wall. At the measurement with ultrasound in B mode of the intimate thickness, the average carotid is a marker of cardiovascular disease(1,2). It has been shown that there is a gradual increase in the cardiovascular risk with the increase in intimate-average carotid thickness, therefore it is important to measure the intima-media carotid thickness and to determine the atherosclerotic plaques in the carotid artery, along with the determination of the classic indicators of FH.
Familial hypercholesterolemia screening (Table 1) should be recommended after the age of 2 years old(5). There are two reasons for that: plasma cholesterol levels are lowest at birth and increase rapidly in the first week of life and then gradually up to 2 years old, when a stagnation of lipid level until the onset of adolescence is observed.
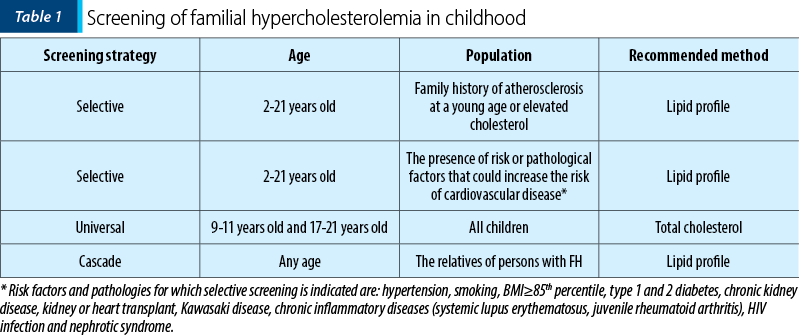
Dietary treatment is not recommended in the literature before 2 years old.
The UK guide recommends screening before the age of 10 years old.
Among the paraclinical investigations (Table 2), the classic lipid profile will be determined, including total cholesterol (CT), triglycerides (TG), LDL-c and HDL-c.
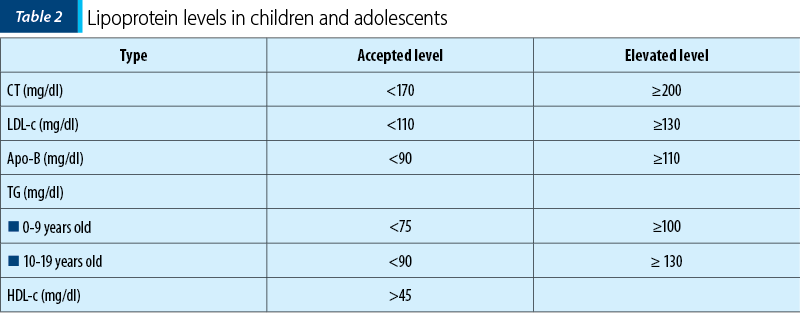
LDL-c should be determined by the Friedewald equation: LDL-c = CT - (HDL-c + TG/2.2). If TG>4.5 mmol/L (or 400 mg/dl), the formula cannot be used and LDL-c must be measured in plasma.
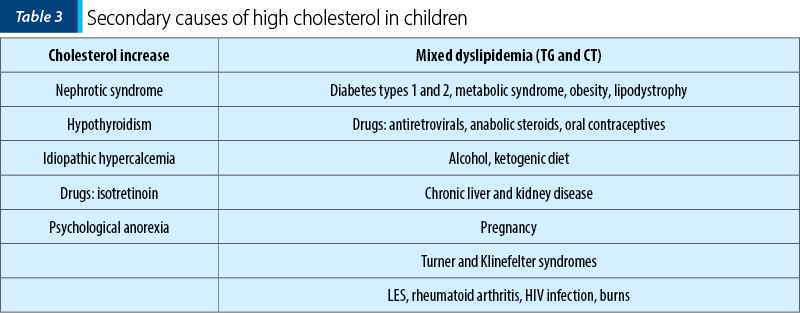
Once the hyperlipidemia is confirmed, it is necessary to exclude some secondary causes of increases elements of the lipid profile (Table 3)(8,9).
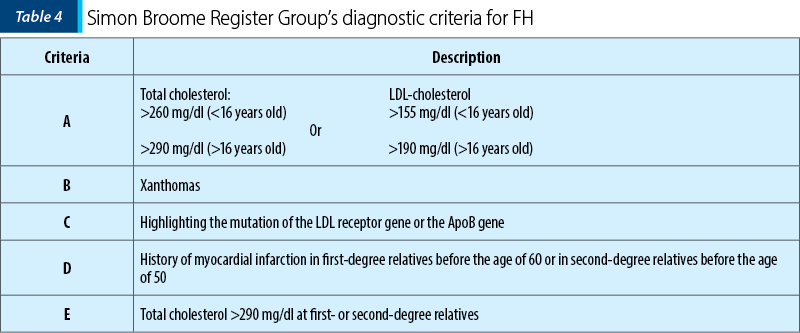
Diagnostic criteria for HF
According to the Simon Broome Register Group’s diagnostic criteria for familial hypercholesterolemia (Table 4), the diagnosis of FH can be: definitive if it meets criteria A and B or C, or a possible diagnostic if criteria A and D or A and E are present.
Regarding molecular diagnosis, the only accepted method for establishing the definitive diagnosis of FH is the determination by molecular tests of mutations in the LDL-r, ApoB or PCSK9 genes. But a negative genetic test result does not rule out the diagnosis of familial hypercholesterolemia(5).
Differential diagnosis
Familial combined hyperlipidemia (FCH) is characterized by elevated levels of CT and LDL-c especially in overweight and obese patients. It is the most common genetic disease with increases in blood lipids causing early coronary heart disease. However, the metabolic and genetic basis of FCH was not fully revealed (a probable cause being represented by decreased LDL-r and increased ApoB).
-
Children with FCH have a high level of CT and/or TG, a low level of HDL-c, with normal values, intermediate or elevated LDL-c.
-
FCH may be concomitant with overweight or obesity and is often associated with insulin resistance.
The treatment of familial hypercholesterolemia in children and adolescents includes: patient’s education, lifestyle and drug treatment.
Patient’s education
Once the diagnosis of FH is established, it is necessary to provide medical care and inform the whole family. The education of the patient and implicitly of the family has become an increasingly appreciated factor since it can play an extremely important role in obtaining positive effects of treatment. The patient needs to know about the causes of familial hypercholesterolemia, the treatment options and the possible complications of the disease(10).
Lifestyle
It is recommended a diet low in saturated fats, avoiding trans fats, and with high fruit consumption, vegetables and fibers. Studies in this regard have shown benefits in the pediatric population. At the same time, changes associated with physical activity may lower LDL-c and improve some disease risk factors like cardiovascular disease in obese children. The information regarding the moment of initiating the dietary treatment or the drugs is presented in Table 5.
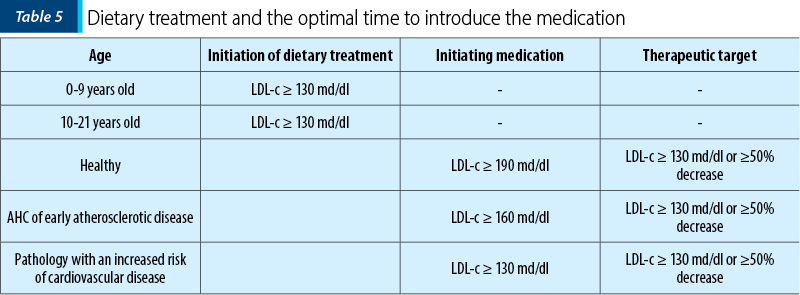
Saturated fatty acids and palmitic acid (the most common fatty acid present in dairy products) reduce LDL receptor expression and increase VLDL synthesis. Instead, unsaturated fatty acids, such as linoleic acid and oleic acid, reduce LDL-c. Reducing saturated fats and cholesterol in children’s diets does not alter the nutritional status, pubertal growth or the development(11,12).
Physical activity
There are studies on the effect of physical activity in other dyslipidemias, but the effect of physical activity on the level LDL-c has not been well studied yet in children with familial hypercholesterolemia. It has been shown that in the pediatric population an increase of physical activity may increase HDL-c and modestly decrease LDL-c. Exercise can also increase insulin sensitivity and help with weight loss(13,14).
Supplements
The most studied supplements used in familial hypercholesterolemia are phytosterol esters and stanol esters. They inhibit the intestinal absorption of cholesterol, causing a 10-15% reduction in LDL-c in children with FH. Studies on omega-3 polyunsaturated fatty acids (alpha linoleic acid, eicosapentaenoic acid and docohexaenoic acid) showed a slight increase in cholesterol levels (mainly due to HDL fraction). Supplementation with omega-3 capsules is not usually recommended in children, it can only be beneficial in certain cases(15,16).
Other supplements mentioned in literature are: soy proteins, cereals, grape seeds, tea, flax seeds, guar gum, garlic, dried fruits etc.
Drug treatment of FH
The introduction of drug therapy in patients diagnosed with familial hypercholesterolemia aims at a reduction in the incidence of coronary heart disease, myocardial infarction, the need to use revascularization procedures and death from cardiovascular cause.
Statins are the first option in FH treatment. Drug treatment is recommended in children after the age of 10 years old, who previously had a specific FH diet for 6-12 months. Studies performed demonstrated that statins are safe in adolescents, without adverse effects on growth and development or on adrenal or gonadal hormones.
Drug treatment is recommended when:
-
LDL-c ≥130 mg/dl in children with diabetes.
-
LDL-c ≥160 mg/dl in children with at least two risk factors for coronary heart disease (obesity, hypertension, smokers) or with a positive family history of early coronary heart disease.
-
LDL-c ≥190 mg/dl in children without risk factors.
Start with low doses of statins (2.5-10 mg/day), gradually increasing the dose. Use in children atorvastatin 10-80 mg/day and rosuvastatin 10-20 mg/day.
The effect of statins on lipid metabolism is observed after 4-6 weeks. Using them as monotherapy does not always target LDL-c levels.
Among the precautions for statin treatment, there are(16,17):
1. Before initiating statin therapy, it is recommended to determine the transaminases and creatinine kinase levels.
2. The level of transaminases should be measured six months after starting the treatment and then at every six months.
3. If the transaminases increase three times or even more than the upper limit of normal, statins should be discontinued. Most of the time, their level normalizes after 2-3 months of discontinuation of statins.
4. The routine determination of creatinine kinase is not recommended during the treatment with statins, but only if muscle symptoms occur (pain and/or muscle weakness). If it exceeds by more than five times the upper limit of normal, it is recommended the interruption of statin treatment.
Contraindications to statins: hypersensitivity to the drug, muscle disorders, myopathy caused by statins, liver disease, persistence of elevated transaminases, values increased more than three times the upper limit of normal during statin treatment, renal failure, severe infections, major surgical conditions, trauma, severe metabolic disorders, hormonal or electrolytic, epileptic seizures.
Conclusions
-
Familial hypercholesterolemia is characterized by an increased level of LDL-c from birth.
-
Vascular changes caused by FH can quickly lead to the development of atherosclerosis, coronary heart disease and even to death.
-
Early diagnosis and proper treatment of HF from childhood can reduce considerable the risk of cardiovascular disease and the sudden death in adults.
-
FH screening is recommended after the age of 2 years old.
-
FH treatment should include educating the patient, an adequate lifestyle and possibly pharmaceutical treatment.
-
Statins are the therapeutic option for familial hypercholesterolemia.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Wiegman A, de Groot E, Hutten BA, Rodenburg J, Gort, J, Bakker HD, Sijbrands EJ, Kastelein JJ, Intimate-arterial mean thickness in heterozygous children for familial hypercholesterolemia. Lancet. 2004;363:369-370.
-
Cheng KS, Mikhailidis DP, Hamilton G, Seifalian AM. An intimate thickness review, carotid and femoral average as an indicator of the presence of peripheral vascular diseases and cardiovascular risk factors. Cardiovascular Res. 2002 Jun;54(3):528-38.
-
De Jongh S, Lilien MR, opt Roodt J, Stroes ES, Bakker HD, Kastelein JJ. Therapy with early statins restore endothelial function in children with hypercholesterolemia family. J Am Coll Cardiol. 2002;40:2117-2121.
-
Perk J, De Backer G, Gohlke H, et al. European guidelines on prevention of cardiovascular diseases in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Euro Heart. 2012 Jul;33(13):1635-701.
-
Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers. 2017 Dec 7;3:17093.
-
Castillo S, Tejedor D, Mozas P, Reyes G, Civeira F, Alonso R, Ros E, Pocovi M, Mata P; on behalf of the Spanish FH Study group. The apolipoprotein B R3500 gene mutation in Spanish subjects with a clinical diagnosis of familial hypercholesterolemia. Atherosclerosis. 2002;165:137-144.
-
Wiegman A, Gidding SS, Watts GF, et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015 Sep 21;36(36):2425-37.
-
Berneson GS, Srinivasan SR, Bao W, et al. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med. 1998 Jun 4;338(23):1650-6.
-
McMahan CA, Gidding SS, Fayad ZA, et al. Risk scores predict atherosclerotic lesion in young people. Arch Intern Med. 2005 Apr 25;165(8):883-90.
-
Noakes TD, Windt J. Evidence that supports the prescription of low- carbohydrate high-fat diets: narrative review. Br J Sports Med. 2017 Jan;51(2):133-9.
-
Siri-Tarino PW, Krauss RM. Diet, lipids, and cardiovascular disease. Curr Opin Lipidol. 2016 Aug;27(4):323-8.
-
Diamond DM, Alabdulgader AA, de Lorgeril M, et al. Dietary Recommendations for Familial Hypercholesterolaemia: an Evidence-Free Zone. BMJ Evidence-Based Medicine. 2020; doi: 10.1136/bmjebm-2020-111412.
-
Stoll M, Dell Oca N. Genetica de la Hypercolesterolemia familiar. Rev Urug Cardiol. 2019;34:324-332.
-
Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J Am Coll Cardiol. 2020 May 26;75(20):2553-2566.
-
Keiji M, Asako M, Hai Ying F, Shohei I, Hayato T,Masa-aki K, Ichrio Y, Tsuyoshi S, Shigeru I, Jun K, Takashi I, Tomohiro H, Katsunori Y, Yoichi H, Takuji F, Kazushige D, Takashi K, Tetsuo M. Universal Screening for Familial Hypercholesterolemia in Children in Kagawa, Japan. Journal of Atherosclerosis and Thrombosis. 2021 Jun 26. doi: 10.5551/jat.62780.
-
Dombalis S, Nash A. The Effect of Statins in Children and Adolescents with Familial Hypercholesterolemia: A Systematic Review. J Pediatr Health Care. 2021 May-Jun;35(3):292-303.
-
Peterson AL, McNeal CJ, Wilson DP. Prevention of Atherosclerotic Cardiovascular Disease in Children with Familial Hypercholesterolemia. Curr Atheroscler Rep. 2021 Aug 27;23(10):64.
Articole din ediţiile anterioare
CASE REPORT | Ediţia 4 64 / 2021
Carcinomul corticosuprarenal – evoluţia fatală a unui tip rar de cancer la copil
Georgiana Scurtu, Magdalena Starcea, Mirabela Alecsa, Silvia Dumitraş, Antonela Ciobanu, Anca Ivanov, Adriana Mocanu, Prof. dr. Ingrith Miron, Roxana Bogos
Carcinomul corticosuprarenal la copii este o tumoră rară, dezvoltată din celulele corticale suprarenale. Un pas foarte important în procesul de d...
03 decembrie 2021
EDITORIAL | Ediţia 2 46 / 2017
Tipuri de relaţii interumane în pediatrie
Prof. dr. Evelina Moraru
Pediatria exercitată ca o profesie de credinţă impune în primul rând pasiune, dar şi exerciţiul perpetuu al alinierii la dinamica adaptării unor no...
12 iunie 2017
CLINICAL STUDIES | Ediţia 1 65 / 2022
Sleep-related breathing disorders in the pediatric patient with Prader-Willi syndrome
Sorina Chindriş, Gabriela Vlad, Ana Maria Daviţoiu, Eugenia Buzoianu, Mirela Iancu, Iulia Ţincu, Ioana Adriana Ghiorghiu, Monica Albert, Andrei Zamfirescu, Doina Pleşca
Sindromul Prader-Willi (PWS) este o boală genetică rară, întâlnită cu o incidenţă de 1:15000 de nou-născuţi, identificată la toate rasele şi car...
12 aprilie 2022
UP-TO-DATE | Ediţia 4 64 / 2021
Rolul alimentaţiei naturale în prevenţia unei probleme mondiale de sănătate publică: obezitatea la vârsta pediatrică
Heidrun Adumitrăchioaiei, Alina-Costina Luca
Obezitatea în rândul copiilor şi adolescenţilor a devenit o problemă globală de sănătate, cu o creştere semnificativă a prevalenţei în ultimii an...
03 decembrie 2021