Background. Pheochromocytoma (PH) is a neuroendocrine tumor with a rare incidence in children, that can have high-risk cardiovascular effects. The symptomatology can be absent, or it can start through paroxysmal hypertension associated with headache, sweating, palpitation and dizziness due to catecholamine secretion. Methodology. We report the case of an 8-year-old patient hospitalized in the “Sf. Maria” Emergency Clinical Hospital for Children, Iaşi, Romania, for headache, sweating and palpitation, with progressive deterioration of the general condition, when blood pressure levels remained high despite antihypertensive medication. Results. After identifying the increased levels of urine and plasma metanephrines, the ultrasound and abdominal CT revealed a tumor, and the diagnosis of bilateral pheochromocytoma and secondary arterial hypertension was established. The pheochromocytoma was histopathologically confirmed intraoperatively. The surgical treatment consisted in the subtotal resection of the tumor, with a favorable postoperative and subsequent evolution. Conclusions. A high index of suspicion of pheochromocytoma
should be raised if the main causes of hypertension in children were excluded. The surgical treatment is the gold standard, leading to a normalization of blood pressure values.
Feocromocitomul la copii – prezentare de caz şi scurtă trecere în revistă a literaturii
Pheochromocytoma in children – case report and short literature review
First published: 21 decembrie 2023
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Pedi.72.4.2023.9272
Abstract
Rezumat
Introducere. Feocromocitomul este o tumoră neuroendocrină cu incidenţă rară la copii, care poate avea efecte cardiovasculare cu risc ridicat. Simptomatologia poate fi absentă sau poate debuta prin hipertensiune paroxistică asociată cu cefalee, transpiraţii, palpitaţii şi ameţeli datorate secreţiei de catecolamine. Metodologie. Raportăm cazul unui pacient în vârstă de 8 ani, internat în Spitalul Clinic de Urgenţă pentru Copii „Sf. Maria”, Iaşi, pentru cefalee, transpiraţii şi palpitaţii, cu deteriorarea progresivă a stării generale, în condiţiile în care valorile tensiunii arteriale au rămas ridicate în ciuda
medicaţiei antihipertensive. Rezultate. După identificarea nivelurilor crescute ale metanefrinelor urinare şi plasmatice, ecografia şi CT-ul abdominal au evidenţiat o tumoră, stabilindu-se diagnosticul de feocromocitom bilateral şi hipertensiune arterială secundară. Feocromocitomul a fost confirmat histopatologic intraoperatoriu. Tratamentul chirurgical a constat în rezecţia subtotală a tumorii, cu o evoluţie postoperatorie şi ulterioară favorabilă. Concluzii. Suspiciunea pentru feocromocitom trebuie ridicată dacă au fost excluse principalele cauze de hipertensiune arterială la copii. Tratamentul chirurgical este standardul de aur, conducând la normalizarea valorilor tensiunii arteriale.
Abbreviations: PH – pheochromocytoma; BP – blood pressure; CT – computed tomography; ECG – electrocardiogram; MEN2 = multiple endocrine neoplasia type 2.
Introduction
Pheochromocytoma is a neuroendocrine tumor secreting catecholamines, that arises from chromaffin cells of the sympathetic nervous system adrenal medulla in 85% of cases or extra-adrenal, in the sympathetic chain, in the carotid body or the Zuckerkland organ(1). In children, the mass is unilateral adrenal in 70% of the cases, and only in 30-40% of the cases it is bilateral or extra-above the kidneys. The etiology is mainly idiopathic, but it can be associated with Hippel-Lindau syndrome, tuberous sclerosis type 1 and multiple endocrine neoplasia (MEN) 2a and 2b neurofibromatosis(2). In 13% of the cases, the origin is malignant(3). It accounts for a range from 0.5% to 2% of all cases of hypertension in children, and it is clinically manifested through severe arterial hypertension associated with palpitation, sweating and severe headache due to the hypersecretion of catecholamines out of the circulation(4).
Case report
The patient G.A., aged 8 years and 11 months old, was admitted for bilateral temporal headache, diaphoresis, diffuse abdominal pain, shortness of breath, dry cough and a whimsical appetite, a symptomatology which started insidiously a week before, in full apparent state of health. He had no significant past medical history, and his family history was insignificant; there were no cases of congenital or genetic illnesses in the family. The physical examination showed an influenced general condition, no fever, he was 121 cm tall and weighed 31 kg, with eye shadows, regular rate and rhythm, no murmurs, his pulse was regular at 90/min, and his blood pressure was 140/100 mmHg, with diffuse tenderness to palpation without rebound, guarding or rigidity. Twenty-four hours after admission, he presented a deteriorating state, with sclerosis, pallor, sweating skin, cold extremities, losing conscience, with tonic-clonic seizures with a duration of 2 minutes; RR=25/min, HR=80/min, and BP=225/165 mmHg. The ECG revealed a regular rhythm, the QRS axis was at +90 degrees, PQ=0.12 s, T negative in aVL, the Lyon-Sokolov index=43 mv, ST over-levelled in the right antecordial derivations, Q/S aspect in V1-V2 – front-septal hypertrophy, QT=0.42 dry-prolonged. The chest X-ray was normal. The abdominal ultrasound revealed on the topography of the right adrenal gland a 3.7/2.7 hyperechogenic mass without a Doppler signal, and the left adrenal gland was difficult to visualize. Identical echocardiography: concentric hypertrophic cardiopathy and diastolic left ventricle dysfunction.
The laboratory findings revealed a systemic inflammatory syndrome, elevated white cell count with neutrophilia, elevated platelets count, ALT=183 U1/l, glucose 273 mg/dl, chlorine 87.8 mmol/l, urinary metanephrines/24 h was 80 Aµg/and 98 Aµg (50-350 Aµg/24 h), urinary normetanephrines/24 h was 4168 and 4.914 (100-600/Aµg/24h); homovanillic acid 14.06 mg/24 h; urinary catecholamines: adrenaline 28 µg/24 h (0.2-10 µg/24 h), noradrenaline 3253.91 µg/24 h; dopamine 203 µg/24 h (65-400 µg/24 h); chromogranin A 370 µg/L; iron compounds 102.4 ng/mL (7-84 ng/mL), specific neuron enolases 19.62 ng/24 h (<8 ng/24 h), direct plasmatic kidney compound 2726 pg/mL.
The native and contrast substance abdominal CT examination revealed a solid well delineated expansive mass on the topography of the external arm of the right adrenal gland of 3.3/2.6/4.2 cm (AP/T/CC) with an intense and homogeneous post-CIV contrast trace (Figures 1, 2 and 3).
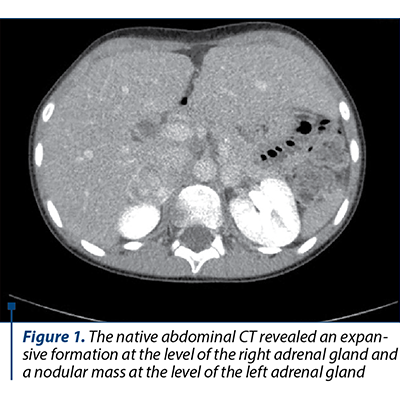
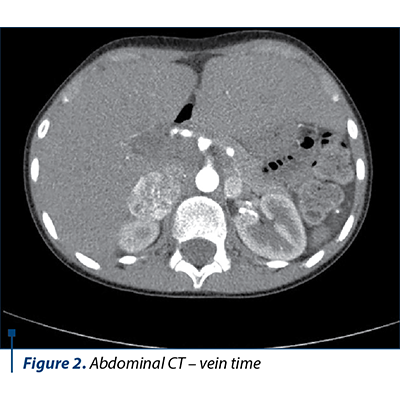
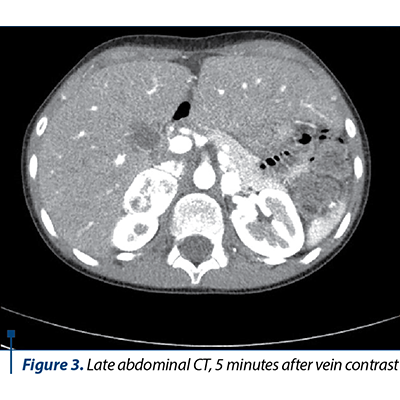
Based on the clinical manifestations represented by severe arterial hypertension associated with headache and profuse sweating, along with the laboratory and medical imaging, the diagnosis of bilateral pheochromocytoma and severe arterial hypertension was established. The differential diagnosis had in mind the exclusion of the main and secondary hypertension causes in children. The initial antihypertensive therapy consisted in the administration of a single antihypertensive with blood pressure (BP) monitoring. Due to unresponsiveness, the successive introduction of antihypertensives was decided upon, as the BP control was stabilized under the scheme: phenoxybenzamine 10 mg/day, amlodipine 5 mg/day, metoprolol 25 mg/day, prazosin 2 mg/day. The subtotal excision of both tumoral masses was performed, and the histopathological result concluded: proliferation of slightly pleomorphous round-oval cells with a finely granular, amphophile cytoplasm and round nucleus with prominent nucleoli, disposed in cubes in a fine fibrovascular stroma; at places, the nuclei are hyperchromic and enlarged in their volume; hemorrhage areas; well delineated by the fibrous capsule in the periphery and adrenal gland tissue in places. After surgery, the evolution was good, without any episodes of hyper- or hypotension, in the absence of medical treatment, with the normalization of the serum and urinary metanephrines, the adrenal cortex function remaining normal.
The short-term diagnosis is favorable, while in the long run, it needs monitoring, with endocrinologic, cardiologic and surgical follow-up.
Discussion
Pheochromocytoma is a tumor with low prevalence within the general population, but especially among the pediatric population, with an incidence of 1 case per 100,000 persons, of which only 10-20% of the cases reported in children. The diagnosis is essential in case of paroxysmal or permanent arterial hypertension with an acute evolution without a specified cause(5,6).
The bilateral pheochromocytoma is associated in 24% of the cases with familiar forms (neurofibromatosis, the von Hippel-Lindau illness, tuberous sclerosis, the Sturge-Weber and MEN2 syndromes). In this case, the family history was negative, the patient needing additional investigations to identify an underlying endocrine pathology(7,8).
Pheochromocytoma is clinically manifested through the classical triad represented by palpitations, paroxysmal attacks of headaches and diaphoresis, as the key to early diagnosis(9). In this case, the appearance of high blood pressure values and seizure led to a full case investigation in order to formulate the diagnosis.
The therapeutic management of arterial hypertension in the pheochromocytoma includes administering alpha- and beta-adrenergic blockers, and lowering BP under the 50th percentile for age and height. The continuous severe arterial hypertension for a period longer than 24 hours is correlated with high levels of plasmatic norepinephrine issued continuously, compared to the tumors secreted by epinephrine(10,11). The cardiac resounding determined by hypertension is revealed by the presence of the ventricular hypertrophy observed at electrocardiograph (the Sokolov-Lyon=43 mV index), and the concentric hypertrophic cardiopathy identified at echocardiography.
Dosing the urinary metanephrines along 24 hours and the plasmatic metanephrines, as well as the characteristic CT aspects offer information regarding the tumoral type and location. The prompt and early discovery of pheochromocytoma is crucial to instituting the antihypertensive medication therapy correlated with the surgical removal of the tumor. In this case, the preoperative medication followed the possibility of a hypertensive paroxysmal crisis in the context of the “catecholamine storm”, which can be precipitated by numerous factors, such as general anesthesia and surgical maneuvers(12,13).
The screening of pheochromocytoma in children is advisable after excluding the causes of secondary arterial hypertension, as well as kidney-related vascular conditions or the aorta coarctation(14,15).
Conclusions
Pheochromocytoma is a rare neuroendocrine tumor which can have major and sometimes fatal cardiovascular effects. The symptomatology varies from case to case, as it is similar both to cardiologic and neurological symptoms. The choice of the antihypertensive therapy is essential to the stabilization of tensional values prior to the surgical intervention.
Acknowledgements: The authors want to thank the Pediatric Cardiology Department and Acute Therapy of the “Sf. Maria” Emergency Clinical Hospital for Children, Iaşi, Romania.
Funding: No funding to declare.
Corresponding author: Alexandrina-Ştefania Curpăn E-mail: andracurpan@yahoo.com
Conflict of interest: none declared.
Financial support: none declared.
This work is permanently accessible online free of charge and published under the CC-BY licence.

Bibliografie
1. Plouin PF, Gimenez-Roqueplo AP. Pheochromocytomas and secreting paragangliomas. Orphanet J Rare Dis. 2006;1:49. Published 2006 Dec 8.
2. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675.
3. Malachias MVB. Feocromocitoma: diagnóstico e tratamento. Rev Bras Hipertens. 2002;9:160–164.
4. Khorram-Manesh A, Ahlman H, Nilsson O, et al. Long-term outcome of a large series of patients surgically treated for pheochromocytoma. J Intern Med. 2005;258(1):55-66.
5. Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am. 2007;21(3):509-ix.
6. Scholz T, Schulz C, Klose S, Lehnert H. Diagnostic management of benign and malignant pheochromocytoma. Exp Clin Endocrinol Diabetes. 2007;115(3):155-159.
7. Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459-1466.
8. Zuber SM, Kantorovich V, Pacak K. Hypertension in pheochromocytoma: characteristics and treatment. Endocrinol Metab Clin North Am. 2011;40(2):295-vii.
9. Shen SJ, Cheng HM, Chiu AW, Chou CW, Chen JY. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung Med J. 2005;28(1):44-50.
10. Bhansali A, Rajput R, Behra A, Rao KL, Khandelwal N, Radotra BD. Childhood sporadic pheochromocytoma: clinical profile and outcome in 19 patients. J Pediatr Endocrinol Metab. 2006;19(5):749-756.
11. National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2 Suppl 4th Report):555-576.
12. Pham TH, Moir C, Thompson GB, et al. Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics. 2006;118(3):1109-1117.
13. Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab. 2007;3(2):92-102.
14. Romero M, Kapur G, Baracco R, Valentini RP, Mattoo TK, Jain A. Treatment of Hypertension in Children With Catecholamine-Secreting Tumors: A Systematic Approach. J Clin Hypertens (Greenwich). 2015;17(9):720-725.
15. Guérin M, Guillemot J, Thouënnon E, et al. Granins and their derived peptides in normal and tumoral chromaffin tissue: Implications for the diagnosis and prognosis of pheochromocytoma. Regul Pept. 2010;165(1):21-29.