Prader-Willi syndrome is a consequence of several genetic defects in the 15q11-q13 region, including methylation changes, and is associated in newborns with hypotonia, poor suck, a reduced growth velocity, and developmental delay. We present the case of a newborn with severe hypotonia delivered via caesarean surgery at full term in the "Elias" University Emergency Hospital. Although the newborn had normal respiratory and cardiovascular parameters, the clinical examination had revealed hypotonia, lethargy and hypogonadism. The risk factors are unknown for this particularly case, as there was no additional risk identified prenatally. The particularity of this case is that the newborn had since birth very severe hypotonia and poor suck.
REVIEW
Sindromul Prader-Willi - prezentare de caz şi revizuirea literaturii
Prader-Willi syndrome - case report and literature review
First published: 24 decembrie 2017
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Peri.1.4.2017.1435
Abstract
Rezumat
Sindromul Prader-Willi este consecinţa mai multor defecte genetice în regiunea 15q11-q13, între care şi modificările de metilare, şi este asociat la nou-născuţi cu hipotonie, dificultăţi de hrănire, creştere redusă şi retard de dezvoltare. Prezentăm cazul unui nou-născut la termen (38 de săptămâni gestaţionale), de sex masculin, născut prin operaţie cezariană în Clinica de Obstetrică-Ginecologie a Spitalului Universitar de Urgenţă „Elias”, cu hipotonie severă. Nou-născutul a avut parametrii respiratori şi cardiovasculari normali, însă examenul clinic a relevat hipotonie severă, reactivitate scăzută şi hipogonadism. Factorii de risc rămân necunoscuţi pentru acest caz, deoarece nu a existat niciun risc suplimentar identificat prenatal. O particularitate a acestui caz este faptul că nou-născutul prezintă de la naştere hipotonie severă şi dificultăţi de alimentaţie.
Introducere
Sindromul Prader-Willi (PWS) este o afecţiune ereditară, care apare prin microdeleţie la nivelul cromozomului 15 patern, în aproximativ 70% dintre cazuri, iar celelalte cazuri sunt datorate disomiei uniparentale sau altor mecanisme incomplet elucidate.
Erorile la amprentarea genomică care apar în timpul gametogenezei masculine şi feminine în sindromul Prader-Willi includ pierderea expresiei genelor paterne, care sunt în mod normal active şi localizate în regiunea cromozomului 15q11-q13(1,2).
Deşi diagnosticul poate fi uşor stabilit, încă din perioada neonatală, datorită caracteristicilor clinice evidente, este necesară şi folosirea metodelor moderne de testare la nivel molecular pentru confirmarea diagnosticului(3).
Prenatal. Diagnosticul prenatal se face rar, dar teoretic ar putea să fie suspectat în cazul mişcărilor fetale reduse şi al polihidramniosului(4). De asemenea, se pot efectua teste genetice obţinute din vilozităţi coriale şi amniocenteză(5). Dimensiunea fetală este în general în limite normale, hipotonia prenatală are ca rezultat, de obicei, scăderea mişcărilor fetale şi poziţia anormală a fătului la naştere, ceea ce duce la creşterea incidenţei de naştere prin operaţie cezariană. Greutatea la naştere şi indicele de masă corporală sunt, în medie, cu 15% mai scăzute decât cele normale.
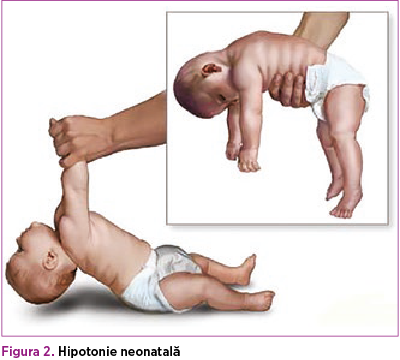
Postnatal. Caracteristicile clinice ale nou-născutului cu sindrom Prader-Willi sunt hipotonia severă observată în mod constant la naştere şi în timpul perioadei neonatale.
Hipotonia din perioada neonatală este asociată cu un plânset slab, letargie şi supt redus, ceea ce necesită tehnici speciale de hrănire.
Alte trăsături ale sindromului Prader-Willi sunt creşterea insuficientă în perioada de sugar, statura mică, letargia, dificultăţi de hrănire, saliva groasă şi raportul circumferinţei cap-piept crescut, hipogonadism atât la bărbaţi, cât şi la femei, cu criptorhidie frecventă la bărbaţi(6,7).
Hipotonia este asociată cu mişcări fetale scăzute, poziţie anormală a fătului şi dificultăţi de mişcare în timpul naşterii. Hipotonia din perioada neonatală este asociată cu un plânset slab, letargie şi supt redus, ceea ce necesită tehnici speciale de hrănire. Drept consecinţă, reflexele sunt afectate şi achiziţiile motorii sunt întârziate. Deşi hipotonia se va îmbunătăţi treptat, adulţii cu PWS rămân uşor hipotonici(3,5).
Manifestările clinice se continuă şi în perioada copilăriei, la copiii netrataţi, incluzând hiperfagie, urmată de obezitate morbidă, creşterea întârziată şi alte caracteristici dismorfice, incluzând bradicefalie, ochii migdalaţi, comisuri bucale cu orientare în jos, mâini şi picioare mici.
Copiii cu sindromul Prader-Willi prezintă tulburări cognitive şi retard mintal slab până la moderat, cu probleme comportamentale frecvent întâlnite, caracterizate prin accese temperamentale, încăpăţânare şi temperament compulsiv.
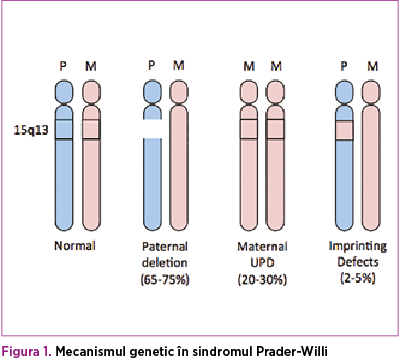
Diagnostic diferenţial. Pacienţii testaţi pentru sindromul Prader-Willi cu rezultate negative trebuie să fie investigaţi şi pentru alte deleţii şi duplicări cromozomiale, precum şi pentru posibile defecte monogenice care sunt asociate caracteristicilor acestui sindrom. Testul FISH poate stabili diagnosticul diferenţiat între sindromul Angelman şi sindromul Prader-Willi, deoarece şi acesta prezintă trăsături asemănătoare, respectiv hipotonie şi dificultăţi de hrănire. Dacă în cazul sindromului Prader-Willi deleţia are cauză paternă, în cazul sindromului Angelman defectul genetic se datorează unei deleţii pe cromozomul 15q11.2-q13 matern.
De asemenea, alte sindroame cu obezitate genetică, cum sunt sindromul Bardet-Biedel şi sindromul X fragil, asociate cu tulburări cognitive, pot provoca uneori confuzii clinice. În cazul sindromului Bardet-Biedel, deşi tulburările de vedere nu se observă, în mod obişnuit, până la vârsta de 6-8 ani, sunt prezente alte trăsături în fenotipuri, cum este polidactilia (două din trei cazuri) sau brahidactilia, care pot stabili diagnosticul diferenţial precoce(8,9,10).
Stabilirea precoce a diagnosticului şi introducerea imediată a terapiei corespunzătoare au dus la reducerea mortalităţii şi prevenirea complicaţiilor postnatale, cu îmbunătăţirea semnificativă a calităţii vieţii.
Managementul sindromului Prader-Willi se realizează toată viaţa şi include sport, dietă şi terapie comportamentală(11,12). La acestea se poate aplica terapia cu hormoni de creştere(13).
Prognosticul nu este cel mai bun, dar în ultimii ani calitatea vieţii acestor pacienţi s-a îmbunătăţit, atât timp cât se poate crea un mediu special adaptat la nevoile lor speciale.
Prezentare de caz
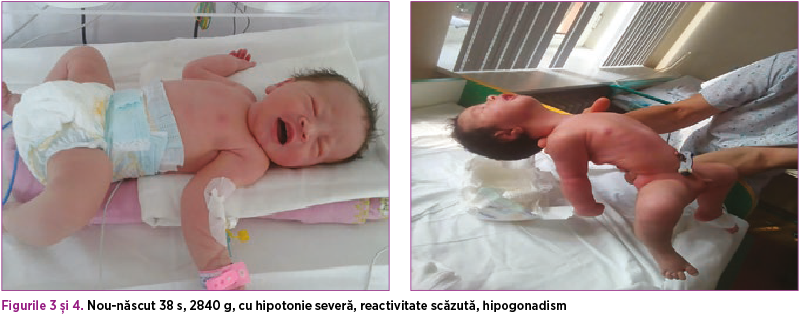
Prezentăm cazul unui nou-născut la termen, AGA, de sex masculin, BIII Rh pozitiv, născut în Clinica de Obstetrică-Ginecologie a Spitalului Universitar de Urgenţă „Elias”, prin operaţie cezariană, prezentaţie craniană, vârsta de gestaţie 38 de săptămâni, scor Apgar 9, greutate la naştere 2840 g, membrane intacte, contracţii uterine dureroase, lichid aminiotic-oligoamnios, cordon ombilical spiralat, provenit din sarcină urmărită şi investigată, cu evoluţie normală, screening infecţios negativ. Mama are 36 de ani, IG IP, grup sangvin BIII Rh pozitiv. Mama afirmă mişcări fetale reduse în sarcină şi polihidramnios. AHC - nepot matern cu celiachie.
La naştere: tegumente rozate, acrocianoză, vernix caseosa, plicatură plantară ½ ant, CO alb sidefiu, 2AA, 1v, craniu osificat, FA-1/1 cm normotensivă, torticolis poziţional dreapta AV>100 bpm, zgomote cardiace ritmice, respiră spontan MV simetric bilateral, cavitatea bucală cu aspect normal, abdomen suplu, OGE de sex masculin, testicule absente în scrot, OGE hipodezvoltate, tonus - hipotonie moderată spre severă, reactivitate scăzută.
Evoluţia în maternitate: nou-născutul este plasat în TINN. Pe perioada internării prezintă hipotonie persistentă, predominant axială, reactivitate prezentă scăzută, ROT prezente, reflex de supt, deglutiţie prezentă. A primit PEV de reechilibrare hidroelectrolitică (70 ml lichide/kgc) şi s-a iniţiat alimentaţie enterală trofică, iniţial prin gavaj, ulterior biberon. A primit antibioterapie profilactică cu ampicilină. Pe parcursul internării evoluţia a fost lent favorabilă, curba ponderală fiind lent ascendentă.
Investigaţii paraclinice: hemograma se menţine în limite normale, AGS: Ph 7,45, pCO 28 mmHg, pO2 - 120 mmHg, FiO2 - 21%, Na 10 mEq/l, K 4,4 mEq/l, Ca 1,25 mmol/l, glucoza 45 mg/dl prereandial, Lac 1,9 mmol/l, HT 45%, BE - 5 mmol/l; glicemii glucotest repetate la valori normale, PCR în dinamică: 15,6 mg/L (valori normale <10 mg/L), CK, CK-MB - în limite normale.
Ecografie transfontanelară - în limite normale.
Consult neurologic: copil hipoton - papuşă de cârpă cu ROT foarte greu de obţinut. Postură de batracian. Tonus scăzut. Forţă scăzută. Reactivitate - trezit cu dificultate din somn, fixează pentru scurt timp, nu plânge, oboseşte la supt, dar suge singur. A fost gavat în primele zile.
Screening metabolic extins (cytogenomic): negativ pentru afecţiunile investigate.
Rezultatul analizei genetice: cariotip 46, xy - cariotip masculin normal.
Rezultatul analizei MS-MLPA a identificat un profil de metilare indicativ pentru sindromul Prader-Willi. Sindromul Prader-Willi este consecinţa mai multor defecte genetice în regiunea 15q11-q13, între care şi modificările de metilare, şi este asociat, la nou-născuţi, cu hipotonie, dificultăţi de hrănire, creştere reduse şi retard de dezvoltare.
Concluzii
Sindromul Prader-Willi este o afecţiune ereditară care apare prin microdeleţie la nivelul cromozomului 15 patern. Caracteristicile clinice ale nou-născutului cu sindrom Prader-Willi sunt hipotonia severă observată în mod constant la naştere şi în timpul perioadei neonatale. Nu sunt citate complicaţii obstetricale specifice în timpul sarcinii, dar este recomandată naşterea prin operaţie cezariană. În majoritatea cazurilor, diagnosticul definitiv depinde de investigaţiile postnatale.
Bibliografie
1. Nicholls RD, Knepper JL (2001) Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet 2:153–175.
2. Goldstone AP (2004) Prader-Willi syndrome: advances in genetics, pathophysiology and treatment. Trends Endocrinol Metab 15:12–20.
3. Bachere N, Diene G, Delagnes V, Molinas C, Moulin P, Tauber M (2008) Early diagnosis and multidisciplinary care reduce the hospitalisation time and duration of tube feeding and prevent early obesity in PWS infants. Horm Res 69:45–52.
4. Whittington JE, Butler JV, Holland AJ (2008) Pre-, peri- and postnatal complications in Prader-Willi syndrome in a U.K. sample. Early Hum Dev 84: 331–336
5. Glenn CC, Deng G, Michaelis RC, Tarleton J, Phelan MC, Surh L, Yang TP, Driscoll DJ (2000) DNA methylation analysis with respect to prenatal diagnosis of the Angelman and Prader-Willi syndromes and imprinting. Prenat Diagn 20:300–306.
6. Cassidy SB. Prader-Willi syndrome. J Med Genet, 1997;34:917–923 (Abstract).
7. Ho AY, Dimitropoulos A. Clinical management of behavioural characteristics of Prader–Willi syndrome. Neuropsychiatr Dis Treat 2010;6:107-118 (Abstract).
8. Gilhuis HJ, van Ravenswaaij CM, Hamel BJ, Gabreels FJ (2000) Interstitial 6q deletion with a Prader-Willi-like phenotype: a new case and review of the literature. Eur J Paediatr Neurol 4:39–43.
9. Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O’Rahilly S, Farooqi IS (2004) A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci 7:1187–1189.
10. Goldstone AP, Beales PL (2008) Genetic obesity syndromes. Front Horm Res 36:37–60.
11. Cassidy SB, McCandless SE: Prader-Willi syndrome; in Cassidy SB, Allanson JE (eds): Management of Genetic Syndromes. Hoboken, NJ: Wiley-Liss, 2005, pp 429–448.
12. Butler MG, Lee PDK, Whitman BY: Management of Prader-Willi Syndrome. Springer: New York, 2006.
13. Aycan Z, Baş VNJ, Prader-Willi syndrome and growth hormone deficiency. Clin Res PediatrEndocrinol. 2014; 6(2):62-7.
2. Goldstone AP (2004) Prader-Willi syndrome: advances in genetics, pathophysiology and treatment. Trends Endocrinol Metab 15:12–20.
3. Bachere N, Diene G, Delagnes V, Molinas C, Moulin P, Tauber M (2008) Early diagnosis and multidisciplinary care reduce the hospitalisation time and duration of tube feeding and prevent early obesity in PWS infants. Horm Res 69:45–52.
4. Whittington JE, Butler JV, Holland AJ (2008) Pre-, peri- and postnatal complications in Prader-Willi syndrome in a U.K. sample. Early Hum Dev 84: 331–336
5. Glenn CC, Deng G, Michaelis RC, Tarleton J, Phelan MC, Surh L, Yang TP, Driscoll DJ (2000) DNA methylation analysis with respect to prenatal diagnosis of the Angelman and Prader-Willi syndromes and imprinting. Prenat Diagn 20:300–306.
6. Cassidy SB. Prader-Willi syndrome. J Med Genet, 1997;34:917–923 (Abstract).
7. Ho AY, Dimitropoulos A. Clinical management of behavioural characteristics of Prader–Willi syndrome. Neuropsychiatr Dis Treat 2010;6:107-118 (Abstract).
8. Gilhuis HJ, van Ravenswaaij CM, Hamel BJ, Gabreels FJ (2000) Interstitial 6q deletion with a Prader-Willi-like phenotype: a new case and review of the literature. Eur J Paediatr Neurol 4:39–43.
9. Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O’Rahilly S, Farooqi IS (2004) A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci 7:1187–1189.
10. Goldstone AP, Beales PL (2008) Genetic obesity syndromes. Front Horm Res 36:37–60.
11. Cassidy SB, McCandless SE: Prader-Willi syndrome; in Cassidy SB, Allanson JE (eds): Management of Genetic Syndromes. Hoboken, NJ: Wiley-Liss, 2005, pp 429–448.
12. Butler MG, Lee PDK, Whitman BY: Management of Prader-Willi Syndrome. Springer: New York, 2006.
13. Aycan Z, Baş VNJ, Prader-Willi syndrome and growth hormone deficiency. Clin Res PediatrEndocrinol. 2014; 6(2):62-7.
Articole din ediţiile anterioare
NEONATOLOGY | Ediţia 1 2 / 2018
Artrogripoza la un nou-născut prematur provenit din sarcină gemelară. Prezentare de caz
Ana Maria Măreşescu, Simona Vlădăreanu, Ana Maria Brădeanu, Adriana Tecuci, Simona Popescu
Artrogripoza congenitală multiplă (AMC) este caracterizată în literatura de specialitate de multiple contracturi articulare congenitale la nou-născ...
07 mai 2018
NEONATOLOGY | Ediţia 3 1 / 2017
Semnificaţia gazelor din cordonul ombilical în encefalopatia nou-născutului – review al literaturii
Otilia Osmulikevici
Studii experimentale şi modele animale au arătat că encefalopatia hipoxic-ischemică este un proces în evoluţie, mai degrabă, decât un eveniment sin...
30 octombrie 2017
REVIEW | Ediţia 3 1 / 2017
Hernie congenitală diafragmatică pe partea dreaptă - prezentare de caz
Gheorghiţa Sardescu, Adriana Sbârcea, Cătălin Cîrstoveanu
Hernia diafragmatică congenitală este o patologie ce constă în existenţa unui defect la nivelul diafragmului abdominal, din pricina căruia are loc ...
30 octombrie 2017
NEONATOLOGY | Ediţia 1 2 / 2018
Tromboză spontană a arterelor femurale la un nou-născut – studiu de caz
Rashmi Kuttysankaran, Sridhar Ramaiah, Otilia Popescu
Tromboza arterială spontană la nou-născut este un eveniment rar întâlnit. De cele mai multe ori este secundară inserţiei de cateter vascular centra...
07 mai 2018