Autism spectrum disorder (ASD) is a very complex neurodevelopmental disorder whose symptomatology has been reported in several different genetic syndromes, one of which is Sotos syndrome. This syndrome is a congenital disorder, inherited autosomal dominant, being caused by a heterozygous pathogenic variant in the NSD1 gene or a deletion of this gene, which are present in more than 90% of individuals with this clinical diagnosis. We describe the case of a 13-year-old patient diagnosed with autism and Sotos syndrome.
Objective. The description of this patient is intended to emphasize the importance of being aware in case of the co-association of ASD with one of the genetic syndromes, in the presence of clinical features not typical for both of them, in order to avoid delays in diagnosis.
Conclusions. The late diagnosis of ASD and Sotos syndrome in our patient also leads us to the conclusion that, in cases of speech and communication delays in children, it is necessary to carry out a careful examination and a detailed assessment of the child’s development, including experts such as pediatricians development, child psychiatrists, geneticists, speech therapists etc. This will help to establish an early diagnosis, which is important for the development of follow-up, coordination of multidisciplinary care, treatment and prognosis.
Late diagnosis of autism spectrum disorder and Sotos syndrome in a child with atypical symptomatology during infancy
Diagnostic tardiv de tulburare de spectru autist şi sindrom Sotos la un pacient cu simptomatologie atipică în copilăria timpurie
First published: 27 aprilie 2023
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Psih.72.1.2023.7931
Abstract
Rezumat
Tulburarea de spectru autist (TSA) reprezintă o disfuncţie de neurodezvoltare foarte complexă a cărei simptomatologie a fost raportată şi în cadrul unor diferite sindroame genetice, unul dintre acestea fiind sindromul Sotos. Acest sindrom este o tulburare congenitală, transmisă autozomal dominant, fiind cauzat de o variantă patogenică heterozigotă la nivelul genei NSD1 ori o deleţie a acestei gene, prezente la mai mult de 90% dintre persoanele cu acest diagnostic clinic. Prezentăm cazul unui pacient de 13 ani, diagnosticat cu autism şi sindrom Sotos.
Obiectiv. Descrierea acestui caz are scopul de a sublinia importanţa faptului de lua în considerare posibila asociere a TSA cu un sindrom genetic, în prezenţa unor trăsături clinice atipice pentru fiecare tulburare, pentru a evita întârzierile în diagnostic.
Concluzii. În cazul pacientului nostru, diagnosticarea tardivă a TSA şi a sindromului Sotos a condus la concluzia că, în cazul unor copii cu întârzieri de vorbire şi comunicare, este necesară efectuarea unei examinări atente şi detaliate, care să includă pediatri specializaţi în dezvoltare, medici specializaţi în psihiatrie infantilă, geneticieni, logopezi etc. Acest fapt va ajuta la stabilirea unui diagnostic precoce, important pentru iniţierea unui program de follow-up, coordonarea îngrijirii multidisciplinare, tratament şi prognostic.
Introduction
Autism spectrum disorder (ASD), with a multifactorial etiology and diagnosis on the rise, is a genetically heterogeneous group of neurobehavioral disorders characterized by impairment of communication, social interaction and stereotypic repetitive behaviors. Its symptomatology has been reported in a number of congenital syndromes(1,2). Currently, genetic syndromes or chromosomal abnormalities such as tiny DNA deletions or duplications, single gene disorders, gene variations, metabolic irregularities and mitochondrial malfunction are detected in up to 40% of people with ASD(1,2).
One of the genetic syndromes associated with ASD is Sotos syndrome (SS), a congenital disorder that was first described in 1964. Sotos syndrome, with an estimated incidence of 1 in 10,000-14,000 births, is transmitted in an autosomal dominant manner and is caused by a heterozygous pathogenic variant in the NSD1 gene or a deletion of this gene that is located at chromosome 5q35(3-7). According to some studies, the NSD1 gene may be mutated in more than 90% of individuals with a clinical diagnosis of Sotos syndrome(4,5). The cardinal clinical features present in more than 90% of patients with Sotos syndrome are distinctive facial features, intellectual disability, prenatal and postnatal overgrowth. Other major features are found in 15-89% of SS cases, while the associated features are met in 2-15% of individuals with this disorder(4-6).
For the first time, a child with Sotos syndrome who fulfilled the diagnostic criteria for ASD was reported by Morrow et al. in 1990(8). The prevalence and symptomatology of ASD in Sotos syndrome have been widely studied, and systematic literature reviews have found a strong correlation between them, adding Sotos syndrome to the list of genetic syndromes associated to ASD(9,10). The largest cohort study of SS patients, which found that autistic symptomatology may be more prevalent in Sotos syndrome than many other genetic syndromes, also came to the suggestion that age affects how severe these patients’ ASD symptoms are(10).
We describe the case of a 13-year-old male patient diagnosed with autism and Sotos syndrome. This paper demonstrates how we should consider the co-association of an autism spectrum disorder with a genetic syndrome in order to avoid delays in the diagnosis of each of their conditions when there are clinical characteristics that are not typical for both of them.
Case report
The patient was born from a normal pregnancy, by caesarean section, with an uneventful postnatal period. He presented a delay in developmental milestones, with language developmental delay (first words spoken around 36 months of age). The poor motor developmntal enabled him to walk independently at around 24 months, and the quality of interaction and play with others turned out to be difficult. At the age of 36 months old, he was diagnosed with third-degree bronchial asthma. At a head CT and after neurosurgery consultation, it was revealed an isolated sinister temporal arachnoid cyst, without the need for surgical treatment.
Since the difficulties in global development were mild, the child was brought to the doctor’s attention at the beginning of school. Thus, at the age of 6.5 years old, after a detailed developmental evaluation, it was noticed the presence of a somewhat more peculiar appearance (macrocephaly, broad forehead and narrow jaw), along with difficulties in the learning process, the inability to maintain attention, and hyperactive behavior. Only around the age of 10, he was diagnosed with mild mental retardation and attention deficit hyperactivity disorder (ADHD), with marked learning difficulties (e.g, he couldn’t read in the fourth grade of elementary school).
At the age of 10-11 years old, the main concern was aggressive behavior in the school environment and self-harm (he scratched and bit the skin of his hands), social isolation and nocturnal enuresis. The neurological examination was normal and he was placed on risperidone at a dose of 1 mg/day.
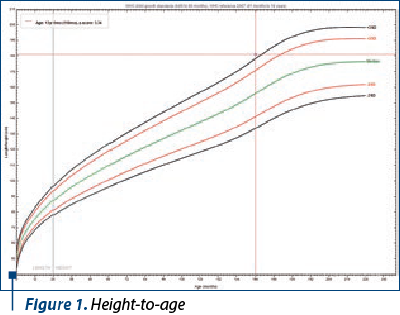
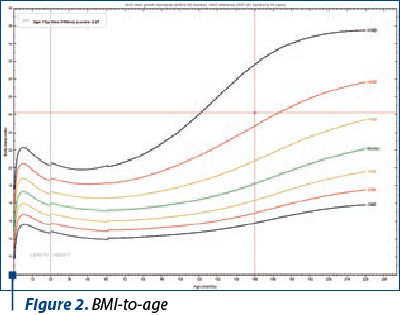
He was first brought to the child and adolescent psychiatrist consultation at the age of 12 for behavioral problems, especially aggressivity and involvement in fighting situations at school. Regarding appearance, the height, macrocephaly, wide forehead and narrow jaw were noticeable. He presented with a physical overgrowth with a height-to-age ratio of over 3 standard deviations (Figure 1) and BMI according to age above 2 standard deviations (Figure 2).
The mental status examination (MSE) highlighted the presence of irritability, inability to concentrate, impulsive behavior up to damaging things, as well as speech with phonetic difficulties. He showed poor speech with difficulties at the morphosyntactic and pragmatic level of the language, without the presence of questions when needed, difficulties in the organization and expression of thought, and an inability to have a spontaneous conversation. Difficulties having a talk with him consisted of his own “way” of speaking with long answers; with many detailed narrative elements and information unrelated to the request; poor working memory, and poor ability to repeat a previously learned action; if he sometimes passed after the instruction, he would forget what it was asked of him. Having difficulties in coordinating actions toward a certain result, in spatial orientation and organizational skills, he couldn’t follow instructions and showed poor assimilation and adaptation of new mental schemes. Often, his answers lacked argumentation and reasoning, were infantile and without using problem-solving techniques/strategies.
Regarding social communication skills, he made eye contact when communicating and even smiled, but he had difficulties engaging in relations with peers or adults. He manifested emotions, managed to talk about things he liked or disliked, and judged well when an action was right or wrong. He had deficits in social-emotional reciprocity, with failure of normal back-and-forth conversation, reduced sharing of interests, emotions or affect, and failure to initiate social interactions. He had difficulties adjusting behavior to suit various social contexts and difficulties in sharing imaginative play/activity. He had highly restricted interests and strong preoccupations with vehicles, a strong desire to take a car ride with his father every day, irritability and anger if they had to be in another place instead of driving. There was evidence of strict and repetitive behaviors even in the early childhood, as it was stated by parents during the diagnostic interview.
The treatment schemes consisted of the use of risperidone up to 4 mg/day combined with acid valproic as a mood stabilizer up to 1200 mg/day, and the symptoms’ management was achieved with difficulty. The developmental assessment concluded that his condition met the criteria for autistic pervasive developmental disorder (ASD) and mild cognitive impairment.
In order to establish the most accurate diagnosis, as well as to make a differential diagnosis with other genetic syndromes, molecular genetic testing was requested in this patient and his parents. This analysis performed at the Gentogene GMBH Laboratory, in Rostock, Germany, enabled the identification of the diagnosis of Sotos syndrome by finding the presence of a heterozygous pathogenic variant c6541T>C p.(Ser2181Pro) (missense mutation) that causes the substitution of the amino acid serine to the amino acid proline in position 2181 of the protein encoded by the NSD1 gene. Also, the genetic analysis performed on the parents of this patient did not find the presence of this pathogenic variant in them.
Discussion
The patient we report in this paper is diagnosed with Sotos syndrome and autism. The largest cohort investigation of Sotos patients indicated that autistic symptomatology may be more common in Sotos syndrome than in many other genetic syndromes, a fact that is important to better understand both autism and this genetic disorder(10).
More than 90% of individuals with Sotos syndrome present characteristics which are known as the “cardinal features”, such as:
A distinctive facial appearance – the facial features, which are most noticeable between 1 and 6 years old, include a broad and prominent forehead with a dolichocephalic head shape, sparse frontotemporal hair, downslanting palpebral fissures, malar flushing, long and narrow face, and long chin. In adulthood, the appearance is still recognizable, but the face is frequently longer and the chin is more prominent(4,6).
Overgrowth – height and/or head circumference are with 2 standard deviations (SD) above the mean. The prenatal onset of overgrowth is present in Sotos syndrome and many affected babies have birth weights above 2 SD above the mean, even though their birth weights are frequently not proportionally increased. Before the age of 10, the afflicted children frequently show rapid linear growth, being mostly significantly taller than their peers, while after the puberty there is some “normalization” of height, so it may not be noticeably above the norm(4,6). According to several research, the limbs’ growth is mostly responsible for the significant height(11). On the other hand, significant macrocephaly is often present in both children and adults with Sotos syndrome(4,6).
Learning disability – early developmental delay, mild-to-severe intellectual impairment. The vast majority of individuals with Sotos syndrome have some degree of learning disability and of intellectual impairment. The degree of impairment is extremely diverse, ranging from occasional cases with normal development to children with very severe learning difficulties who need life-long care. Children with Sotos syndrome typically have learning disabilities that are characterized by relative verbal and visuospatial memory strength, but relative nonverbal reasoning and mathematical reasoning weakness(12).
Patients with Sotos syndrome of all ages are reported to have a wide range of behavioral problems, such as aggression, phobias and autism spectrum disorder, while attention-deficit/hyperactivity disorder is not common among them(6,9,10-13).
In addition to behavioral problems, major features presented in about 15-89% of Sotos syndrome cases also include advanced bone age, cardiac abnormalities, cranial MRI/CT abnormalities, joint hyperlaxity with or without pes planus, maternal preeclampsia, neonatal complications, renal abnormalities, scoliosis and seizures(4,6).
Our patient’s facial features did not point to a classic Sotos syndrome in infancy. Based on the anamnestic data, despite delayed milestones in the first three years, he had developmental progress throughout the preschool years which did not lead to the need for a detailed assessment of the development of those areas affected by autism spectrum disorder, although the presence of his self-injurious and aggressive behavior could not be explained consistently with ADHD and learning problems.
A detailed developmental evaluation of our patient at the age of 6.5 years old noticed his difficulties in the learning process, his inability to maintain attention and his hyperactive behavior, as well as some facial features such as macrocephaly and broad forehead. At the time, a probable genetic syndrome was supposed, although no genetic testing was performed.
It is important to note the fact that, prior to consulting a child and adolescent psychiatrist at age 12 for his behavioral concerns, our patient was examined by a number of pediatricians and neurologists. And when he was around 10 years old, he was diagnosed with mild mental retardation and attention deficit hyperactivity disorder with marked learning difficulties, without any doubt for ASD symptomatology. He was treated with risperidone at a dose of 1 mg/day for a period of four months. The first consultation with a child and adolescent psychiatrist for his behavioral issues was when the patient was 12 years old. The mental status examination during this child psychiatrist visit highlighted the presence of irritability, inability to concentrate, impulsive behavior up to damaging things, as well as speech with phonetic difficulties. The developmental assessment concluded that his condition met the criteria for autistic pervasive developmental disorder and mild cognitive impairment. Our attention is also drawn to his height, macrocephaly, broad forehead and narrow jaw. Unfortunately, the genetic analysis was requested for the first time when our patient was 12 years old. The molecular analysis revealed the presence of a heterozygous, de novo, pathogenic variant in NSD1 gene, c6541T>C p.(Ser2181Pro) that substitutes aminoacid proline to serine. De novo status has also been confirmed by parental testing, as neither parent of the patient had the NSD1 pathogenic variant. By finding a heterozygous NSD1 pathogenic variant in molecular genetic testing, the diagnosis of Sotos syndrome was established in our patient(6). It was reported that intragenic NSD1 mutations and 5q35 microdeletions encompassing NSD1 are present in at least 90% of Sotos syndrome patients and that more than 95% of individuals with SS have a de novo pathogenic variant of this gene(4-6,14).
It is essential to note that the expressivity in Sotos syndrome is highly variable, meaning that individuals who carry the same pathogenic variation, even within the same family, may have different clinical features(6). As with our patient, who did not show the typical SS features during his infancy, it should be highlighted that in the case of a patient with rapid growth, learning difficulties, macrocephaly, speech delay and schematic behavioral patterns, Sotos syndrome can be considered in the prediagnostic stage(6,15).
It is also important to mention that most people with Sotos syndrome demonstrate clinically significant behavioral symptoms associated with ASD, and their gravity appears to vary with age, with more noticeable behavioral characteristics throughout childhood (ages 5 to 19) compared to early childhood (ages 2.5 to 5) and adulthood (20 years of age and older)(10). The same evolution happened with our patient: at the age range of 3-6 years old, there was progress in global development, he developed expressive language and improved communication, although with qualitative impairment of communication and social interaction. The symptoms began to develop with learning difficulties and then behavioral problems throughout the early school years. Because there may still be cases that are undiagnosed, clinicians must carefully assess ASD in patients with Sotos syndrome and vice versa in the context of co-occurring ASD symptomatology with SS(9,10,13).
The early diagnosis of both syndromes is important for the follow-up and multidisciplinary care coordination(15). In the case of our described patient, we think that the late diagnosis of ASD makes it necessary to carry out a careful examination by teams that include experts from several areas of child development. Since pediatricians are the first who consult children for any problem, they must keep in mind that, if there is evidence of speech and communication delays, it is best to refer all such children for detailed development evaluation by specialists such as developmental pediatricians, child psychiatrists, geneticists, speech therapists etc.
Although a genetic syndrome was suspected when our patient was younger, the genetic testing was performed later. Recent developments in clinical and molecular genetics have improved the diagnosis and our knowledge on the genetic disorders linked to ASD, which helps in the early diagnosis that is very important for the follow-up, coordinating multidisciplinary care and treatment, as well as the prognosis. n
Abbreviations: ASD – autism spectrum disorder; ADHD – attention deficit hyperactivity disorder;
CT – computed tomography; DNA – deoxyribonucleic acid; MRI – magnetic resonance imaging; MSE – mental status examination; SS – Sotos syndrome.
Acknowledgments. The authors thank and acknowledge the patient and his family. Also, the authors would like to acknowledge the Albanian Society of Rare Diseases.
Conflict of interest: none declared
Financial support: none declared
This work is permanently accessible online free of charge and published under the CC-BY.

Bibliografie
-
Genovese A, Butler MG. Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD). Int J Mol Sci. 2020;21(13):4726. doi: 10.3390/ijms21134726.
-
Gyawali S, Patra BN. Autism spectrum disorder: Trends in research exploring etiopathogenesis. Psychiatry Clin Neurosci. 2019;73(8):466-475.
-
Sotos JF, Dodge PR, Muirhead D, Crawfort JD, Talbot NB. Cerebral gigantism in childhood: a syndrome of excessively rapid growth and acromegalic features and a non-progressive neurologic disorder. N Engl J Med. 1964;271:109-16. doi: 10.1056/NEJM196407162710301.
-
Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, Waggoner D, Turkmen S, Cormier-Daire V, Irrthum A, Rahman N; Childhood Overgrowth Collaboration. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet. 2005;77(2):193-204. doi: 10.1086/432082.
-
Tatton-Brown K, Rahman N. Sotos syndrome. Eur J Hum Genet. 2007;15(3):264-71. doi: 10.1038/sj.ejhg.5201686.
-
Tatton-Brown K, Cole TRP, Rahman N. Sotos Syndrome GeneReviews® [Internet]. Last Update: August 1, 2019.
-
Brennan K, Zheng H, Fahrner JA, Shin JH, Gentles AJ, Schaefer B, Sunwoo JB, Bernstein JA, Gevaert O. NSD1 mutations deregulate transcription and DNA methylation of bivalent developmental genes in Sotos syndrome. Hum Mol Genet. 2022;31(13):2164-2184. doi: 10.1093/hmg/ddac026.
-
Morrow JD, Whitman BY, Accardo PJ. Autistic disorder in Sotos syndrome: a case report. Eur J Pediatr. 1990;149(8):567-9. doi: 10.1007/BF01957694.
-
Sheth K, Moss J, Hyland S, Stinton C, Cole T, Oliver C. The behavioral characteristics of Sotos syndrome. Am J Med Genet A. 2015;167A(12):2945-56. doi: 10.1002/ajmg.a.37373.
-
Lane C, Milne E, Freeth M. Characteristics of Autism Spectrum Disorder in Sotos Syndrome. J Autism Dev Disord. 2017;47(1):135-143. doi: 10.1007/s10803-016-2941-z.
-
de Boer L, le Cessie S, Wit JM. Auxological data in patients clinically suspected of Sotos syndrome with NSD1 gene alterations. Acta Paediatr. 2005;94(8):1142-4. doi: 10.1111/j.1651-2227.2005.tb02059.x.
-
Lane C, Milne E, Freeth M. The cognitive profile of Sotos syndrome.
-
J Neuropsychol. 2019;13(2):240-252. doi: 10.1111/jnp.12146.
-
Lane C, Milne E, Freeth M. Cognition and Behaviour in Sotos Syndrome: A Systematic Review. PLoS One. 2016;11(2):e0149189.
-
Donnelly DE, Turnpenny P, McConnell VPM. Phenotypic variability in a three-generation Northern Irish family with Sotos syndrome. Clin Dysmorphol. 2011;20(4):175-181. doi: 10.1097/MCD.0b013e328349182d.
-
Verma A, Salehi P, Hing A, Curda Roberts AJ. Sotos syndrome with a novel mutation in the NSD1 gene associated with congenital hypothyroidism.
-
Int J Pediatr Adolesc Med. 2021;8(3):191-194. doi: 10.1016/j.ijpam.2020.06.001.