The evolution of research in the field of antidepressant pharmacology registered a period of apparent stagnation after the appearance of multimodal agents (vilazodone, approved by FDA in 2011, and vortioxetine, approved in 2013) in the clinical use, which raised concern among mental health specialists, mainly due to the high rates of partial response or nonresponse in patients with major depressive disorder (MDD). Also, the reduced tolerability of some antidepressants, with an important incidence of adverse effects such as weight gain, sexual dysfunctions or sedation, supported the need for further pharmacological explorations in order to find new therapeutic solutions in MDD. The “new wave” of antidepressants includes agents with different mechanisms of action, namely partial agonism of type 1A serotoninergic receptors, glutamatergic modulation, simultaneous inhibition of the three monoamine transporters, allosteric modulation of GABA-A receptors, or the combination of distinct mechanisms of several pharmacological agents in a single tablet. Gepirone, esketamine, toludesvenlafaxine, zuranolone, brexanolone and the combination dextromethorphan+bupropion are the representatives of the new generation of antidepressants, all of which have been approved in the last five years by various national or regional agencies with responsibilities in the field of medicines authorization, for depressive disorders (MDD, postpartum depression, or treatment-resistant depression). These new agents came not only with the promise of better efficacy (esketamine, dextromethorphan+bupropion) or indications for which there were no specific treatments until now (brexanolone), but also of a superior tolerability profile (gepirone and toludesvenlafaxine). Given that the number of studies supporting the approval of these drugs is currently limited, pharmacovigilance activity and new independent studies will determine whether we can talk about a therapeutic paradigm shift in depression.
Noua generaţie de antidepresive: sunt aceşti agenţi farmacologici capabili să schimbe paradigma terapeutică în tulburarea depresivă majoră?
The “new wave” of antidepressants: are these agents paradigm-shifters in treating major depression?
First published: 29 noiembrie 2023
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Psih.75.4.2023.8926
Abstract
Rezumat
Evoluţia cercetării în domeniul farmacologiei antidepresivelor a înregistrat o perioadă de aparentă stagnare după apariţia în uz clinic a agenţilor multimodali (vilazodona, aprobată de FDA în anul 2011, şi vortioxetina, aprobată în 2013), suscitând îngrijorare în rândul specialiştilor în sănătate mintală, în principal din cauza ratelor crescute de răspuns parţial sau lipsă de răspuns în cazul pacienţilor cu tulburare depresivă majoră (TDM). De asemenea, tolerabilitatea redusă a unor agenţi farmacologici cu proprietăţi antidepresive, manifestată prin efecte adverse precum creşterea în greutate, disfuncţii sexuale ori sedare, indică nevoia continuării cercetărilor în vederea găsirii de noi soluţii terapeutice. „Noul val” de antidepresive include agenţi cu mecanisme de acţiune de tipul agonismului parţial al receptorilor serotoninergici de tip 1A, modulării glutamatergice, inhibiţiei recaptării concomitente a celor trei transportori de monoamine, modulării alosterice a receptorilor GABA-A, ori combinarea unor mecanisme distincte ale mai multor agenţi farmacologici într-o singură tabletă. Gepirona, esketamina, toludesvenlafaxina, zuranolona, brexanolona şi asocierea dextrometorfan+bupropion sunt reprezentanţii noii generaţii de antidepresive, fiind aprobaţi în ultimii cinci ani de diferite agenţii naţionale sau regionale cu responsabilităţi în domeniul autorizării medicamentelor, pentru indicaţii din spectrul depresiei (depresia majoră, depresia post-partum sau depresia rezistentă la tratament). Aceste noi medicamente vin nu doar cu promisiunea unei eficacităţi mai bune (esketamina, dextrometorfan+bupropion) sau a unor indicaţii pentru care nu existau până acum tratamente specifice (brexanolona), ci şi a unui profil de tolerabilitate superior (gepirona şi toludesvenlafaxina). Având în vedere că dovezile care sprijină utilizarea acestor medicamente sunt deocamdată limitate, activitatea de farmacovigilenţă şi viitoarele studii clinice independente vor fi cele care vor determina dacă se poate vorbi despre o schimbare de paradigmă terapeutică în tulburările depresive.
Introduction
The partial response and treatment resistance in major depressive disorder (MDD) is a sad reality with a strong negative impact on social functionality in adult patients. About 20-40% of the patients receiving treatment for MDD or bipolar depression diagnosis have an insufficient clinical response(1-4). A symptom-free status is an aim difficult to achieve in patients with bipolar or monopolar depressive episodes, and only about 33% can achieve full remission(3). Residual symptoms, like depressed mood, anxiety, sleep dysfunctions or fatigue, are powerful predictors of relapse (up to 3-6 times higher versus patients who achieve complete remission), and these symptoms may also be associated with more medical and psychiatric visits, disability benefits, chronic evolution, risk of stroke and coronary events, and even suicidal attempts(3).
Considerable efforts have been invested in finding new therapeutic agents for MDD and bipolar depression, and agents from extremely different pharmacological classes have been subjected to intense scrutiny in preclinical and clinical studies. Many antidepressants in the pipeline are based on the monoaminergic hypothesis, but there are also other mechanisms explored by researchers, such as glutamate neurotransmission, GABA-A receptor modulation or sestrin modulation, and agents acting as cholinergic receptor ligands or orexin receptor antagonists, anti-inflammatory drugs etc.(5-8)
The characteristics of the “new wave” of antidepressants are explored in this paper, based on a literature search that involved querying electronic databases (Google Scholar, PubMed and Clarivate/Web of Science) and gray literature (data from manufacturers, press releases etc.). Only antidepressants approved for clinical use in the last five years were allowed for further analysis.
The “new wave” of antidepressants
A number of six pharmacological agents or drug combinations approved for treating depressive episodes were found: gepirone, bupropion+dextromethorphan, zuranolone, brexanolone, esketamine, and toludesvenlafaxine. While all of them target depressive symptoms, specific indications also exist, such as postpartum depression for brexanolone and treatment-resistant major depression for esketamine.
Gepirone is a new serotonin modulator antidepressant with selective 5HT1A agonist properties, which makes it a closely related agent to buspirone, both being classified within the chemical class of azapirones (Figure 1)(9). However, gepirone does not possess in vitro dopamine receptor binding and has a higher bioavailability than buspirone(9). The potency of 5HT1A receptor binding is presumably higher for gepirone than for selective serotonin reuptake inhibitors (SSRIs), and its action at pre- and post-synaptic serotonin receptors is different (i.e., agonist and partial agonist, respectively), as proven by long-term studies(10-12). Gepirone can also down-regulate 5HT2 receptors, without a significant impact on b-adrenergic receptors(9). This receptoral profile makes it beneficial when sexual dysfunction or weight gain as adverse effects should be avoided, because in clinical trials these events were similar in the active drug and the placebo groups(13).

Gepirone extended-release (ER) was approved by the U.S. Food and Drug Administration (FDA) in September 2023 for the treatment of major depressive disorder in adults(14), but there are no available data about the submission for this drug’s approval from the European Medicines Agency (EMA). Gepirone is also explored for other disorders, like cocaine dependence or anxiety disorders(10,15).
No results have been disclosed on a phase II clinical trial that enrolled 41 patients with cocaine use disorder(15). However, in a randomized, double-blind clinical trial that enrolled patients with generalized anxiety disorder (GAD), gepirone has proven to have an anti-anxiety effect(10). In this trial, which compared gepirone (mean daily dose of 19±11.5 mg/day) and diazepam (19.5±12.5 mg/day) during eight weeks, the 5HT1A receptor agonist had delayed anxiolytic effect compared to diazepam (six weeks versus one week), but the therapeutic activity was significant(10). The main adverse events reported by patients treated with gepirone were lightheadedness, nausea and insomnia(10). Another small, open-label, six-week study investigated the effects of gepirone (mean maximal dose 41 mg/day) on GAD core symptoms and concluded that Hamilton Anxiety Rating Scale (HAMA) scores improved significantly (p<0.001)(16).
According to an older clinical trial (N=130 patients), gepirone administered in a 5-30 mg/day dose range led to superior effects on depressive symptoms, including melancholic manifestations, during an eight-week period of monitoring(9). Also, in the four-week continuation trial that enrolled patients (N=137) with an initial response to six-week open therapy with gepirone, the Hamilton Depression Rating Scale (HAMD) scores decreased significantly(9). Yet another trial, that included 209 patients with moderate-to-severe MDD, showed positive results for gepirone versus placebo, as determined by the HAM-17 scores evolution during the eight weeks of active monitoring(17). The risk of weight gain and sedation was not different between the gepirone and placebo-treated patients(17).
The effect of gepirone ER on sexual function in patients with MDD was explored by a clinical trial that compared this drug with fluoxetine and placebo in 181 male patients(18). After eight weeks, gepirone ER improved total sexual function versus placebo, especially the “orgasm” domain; even patients who were nonresponders to this drug’s antidepressant and anxiolytic effects still showed significant improvement in sexual function, unlike fluoxetine(18).
A similarly favorable impact on sexual function, independent of antidepressant/anxiolytic activity, was reported by a post hoc analysis in women with MDD who received gepirone, reflected by an improvement of “sexual desire” in the short-term and long-term(19). Even more, a post hoc analysis that explored specifically the effect of gepirone on women with hypoactive sexual desire disorder (HSDD) and MDD (N=161 participants; 18 to 64 years old) supported a significant favorable effect of the 5HT1A receptor agonist versus placebo (63% HSDD improvement versus 40% after eight weeks)(20).
A review of clinical trials with gepirone showed that gepirone immediate-release (IR) had significant effects on depressive symptoms, but due to its short half-life, it necessitated frequent administration, and higher peak plasma concentrations were reported (leading to adverse events)(21). Therefore, the ER formulation was conceived as a way to decrease the rate of adverse events, by reducing the peak-to-trough fluctuations in plasma concentration; due to this formulation, higher daily doses of gepirone could be used(21).
The pharmacokinetics of gepirone refer to an absolute bioavailability of 14% to 17%, Cmax reached after 6 hours, Tmax decreased to 3 hours after a high-fat meal, steady-state achieved in 2-4 days, the volume of distribution was 94.5 l, plasma protein binding was 72%, a mean terminal half-life of 5 hours, metabolization by CYP3A4 to two major metabolites that are pharmacologically active (1-PP and 3’-OH-gepirone), and excretion through urine (81%) and feces (13%)(22,23). Hepatic or renal impairment negatively impacts the clearance of gepirone, therefore the maximal dose should be limited to 36.3 mg/day in these patients(22).
The dosing regimen recommended is 18.2 mg orally once a day, taken with food, and the dose should be titrated, depending on the response, to 36.3 mg after four days, and it could be further raised to 54.5 mg/day (day 7) and 72.6 mg/day (day 14)(22). When co-administered with CYP3A4 inhibitors, the dose of gepirone should be reduced by 50%(22). In the geriatric population, the dose may be raised up to 36.3 mg/day after seven days(22). The warnings included in the FDA-approved Summary of Product Characteristics (SPC) are QT interval prolongation, serotonin syndrome, and activation of mania/hypomania(22).
The FDA approved the combination of bupropion hydrochloride and dextromethorphan hydrobromide in August 2022 as an oral, fixed-dose association for treating MDD in adults(24,25). Other indications for this combination that are explored include agitation related to Alzheimer’s disease, and nicotine use disorder(25). While dextromethorphan is an uncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist and s1 receptor agonist, bupropion is a norepinephrine and dopamine reuptake inhibitor with CYP2D6 inhibition properties(24). Also, the metabolites of bupropion are CYP2D6 inhibitors, a pharmacologic property that leads to higher dextromethorphan plasma levels, as confirmed by three phase I studies(26). This pharmacokinetic profile is important because dextromethorphan is rapidly metabolized by cytochrome P450 and the therapeutic plasma levels are difficult to obtain after its oral administration(26).
From a pharmacodynamic perspective, both drugs enhance the norepinephrine neurotransmission by reuptake inhibition and a4b2 nicotinic receptor antagonism, indicating a synergistic activity(26). Besides the aforementioned properties of dextromethorphan, it should also be mentioned its effect on serotonin neurotransmission via s1 agonism in the dorsal raphe nuclei(26). It was presumed that the dextromethorphan/bupropion combo would be more efficient and would induce a more rapid effect than antidepressant monotherapy, due to its multimodal and multitarget properties(26).
A phase III, double-blind clinical trial (N=327 patients with MDD) concluded that dextromethorphan/bupropion combination was superior to the placebo in decreasing the MADRS scores at week 6, with a rate of remission reaching 39.5% versus 17.3% at endpoint(27). The most common adverse events reported in patients treated with the active drug were dizziness, nausea, somnolence, dry mouth and headache(27). A randomized, double-blind, multicenter, parallel-group trial evaluated dextromethorphan/bupropion (45/105 mg) versus bupropion SR (105 mg) in 97 patients with MDD during six weeks; the end-point results showed a significantly greater MADRS score reduction in the first group versus the bupropion-only treated patients(28). Response rates and remission rates were also higher in patients treated with the combination of dextromethorphan and bupropion(28). Another phase III study (STRIDE-1, N=312 patients with MDD) achieved only secondary efficacy endpoints for dextromethorphan/bupropion (45/105 mg x 2/day) at weeks 1, 2, and 6 (MADRS scores) versus active comparator (150 x 2 mg/day)(29).
According to a recent literature review (n=9 papers) that included preclinical and clinical data on dextromethorphan, this pharmacological agent is “well tolerated and exhibits clinically significant effects”(30). The abuse potential or the gateway effect of the dextromethorphan/bupropion combo has not been systematically assessed(31), therefore caution is still recommended for vulnerable populations. The rapid conversion of dextromethorphan to dextrorphan is considered a potential risk factor for the abusive use of this drug, and preclinical studies in rodents and monkeys showed an abuse potential for both the parent drug and its active metabolite(32,33). A systematic review (n=5 studies with a low risk of bias) supported the efficacy of dextromethorphan/bupropion in decreasing the MADRS scores as early as seven days after the baseline versus placebo and after two weeks when compared to an active treatment(34). The treatment efficacy was maintained for up to one year, and this phenomenon was confirmed by clinical remission and response rates(34). The adverse events reported were transient(34). Regarding the pivotal studies and other studies that supported the approval of the dextromethorphan/bupropion combo, a potential risk for “sponsor and investigator biases” could not be excluded, because four preclinical studies and two clinical trials (out of nine sources) were sponsored by the same entities(31).
Case reports about the efficacy of the dextromethorphan/bupropion combination also exist. In such a report, a 42-year-old female patient diagnosed with severe, treatment-resistant MDD, posttraumatic disorder and borderline personality disorder was initiated on bupropion (450 mg/day) and trazodone (200 mg/day), but this treatment showed only partial effectiveness, therefore repetitive transcranial magnetic stimulation therapy was carried out, but the response was incomplete(35). The patient attempted suicide five weeks after admission, and the case manager decided to initiate the treatment with dextromethorphan/bupropion (30/450 mg), reducing the dose of bupropion to 150 mg/day because its serum level was too high(35). The effect was favorable, with a significant decrease in depressive symptomatology and suicidal behavior starting from two weeks of treatment(35).
Although initially patented for its anti-tussive properties in 1949, the research of dextromethorphan’s properties evolved rapidly and it has been repurposed as an antidepressant, but it is also explored for the treatment of stroke, traumatic brain injury, pain etc.(31,36) The FDA approved the combination of dextromethorphan and quinidine for treating the pseudobulbar affect in adults; EMA also approved this combination of drugs, but in February 2016 the authorization was withdrawn at the manufacturer’s request(30,37). A small proof-of-concept clinical trial (N=20 participants) explored the efficacy and tolerability of dextromethorphan/quinidine (45/10 mg/12 h) during 10 weeks(38). This open-label phase II trial showed a decrease in the Montgomery-Asberg Depression Rating Scale (MADRS) and Quick Inventory of Depressive Symptomatology (QIDS-SR) scores at a significant level(38). The remission rate according to the MADRS scores was 45%(38). There are also reports on the favorable effects of dextromethorphan in bipolar depression(31,36). The association of dextromethorphan and memantine as an add-on to valproate has been efficient in patients with depressive episodes within type II bipolar disorder, in a 12-week randomized, placebo-controlled clinical trial(31).
The warnings included in the package leaflet include a dose-related risk of seizure, high blood pressure, activation of mania/hypomania, psychotic reactions, angle-closure glaucoma new onset, dizziness, serotonin syndrome, and embryo-fetal toxicity (Figure 2)(24). Contraindications for the administration of dextromethorphan/bupropion combo are represented by seizure disorders, current or prior diagnosis of anorexia nervosa, withdrawal of alcohol, benzodiazepines, barbiturates and antiepileptics, concomitant use with monoamine oxidase inhibitors (MAOIs), and hypersensitivity to the components of this drug(24).
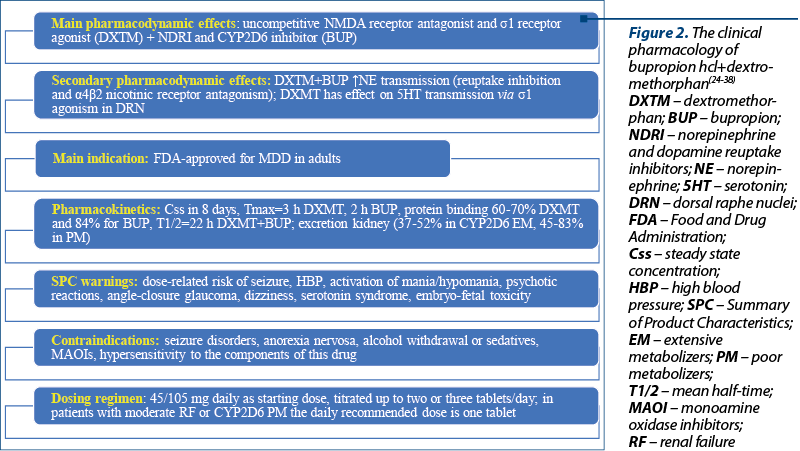
The starting dose is one tablet (45/105 mg dextromethorphan hydrobromide/bupropion hydrochloride) daily, and the increase to the maximum recommended dose (two tablets/day) could be done after three days(24). In patients with moderate renal impairment and CYP2D6 poor metabolizers, the maximum daily dose is one tablet(24).
The steady-state plasma concentration of dextromethorphan/bupropion is reached within eight days, the median Tmax is 3 hours for dextromethorphan and 2 hours for bupropion, and the effect of food on the drug’s absorption is negligible(24). The plasma protein binding of dextromethorphan is 60-70%, and for bupropion – 84%; the mean elimination half-life of dextromethorphan increases to 22 hours when administered with bupropion, which is three-fold higher than the monotherapy; the mean elimination half-life of bupropion is 15 hours(24).
The excretion of dextromethorphan is mainly through urine (37-52% in extensive CYP2D6 metabolizers, and 45-83% in poor metabolizers)(24). There are no specifications about the pharmacokinetics of dextromethorphan/bupropion association in geriatric or pediatric populations(24).
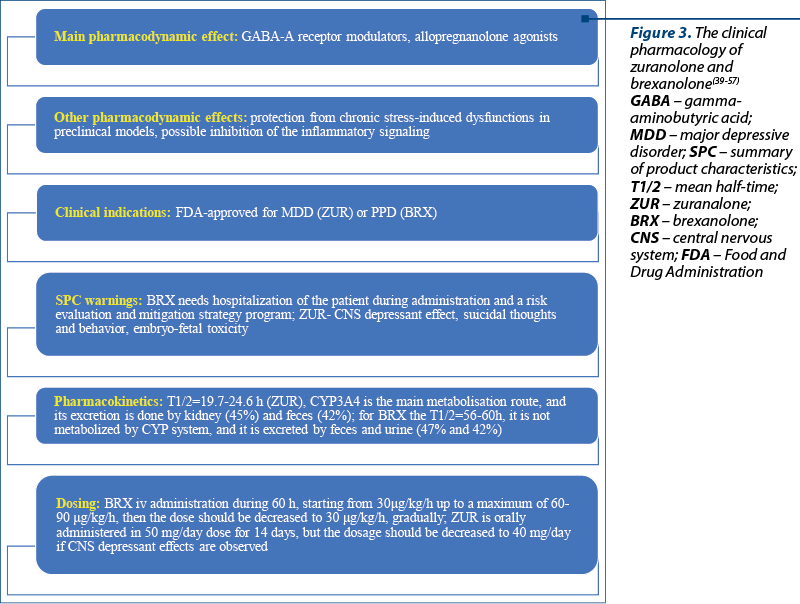
Zuranolone and brexanolone target g-aminobutyric acid (GABA) neurotransmission as an allosteric modulator of GABA-A receptors, and are classified as “allopregnanolone agonists”(39,40). The rapid therapeutic effect of these neurosteroid analogs was not previously reported by any other antidepressant, which may represent a clear advantage for managing major depression, since most antidepressants do not associate therapeutic response before two weeks(41). FDA approved brexanolone in March 2019 for treating PPD and zuranolone for MDD in August 2023(42,43).
Neurosteroids were explored as components of the excitatory-inhibitory neuronal balance and modulation of the stress reactions(41). In vitro studies showed that zuranolone may enhance the GABA-A receptor current in presynaptic and postsynaptic configurations (g subunit and d subunit-containing configurations, respectively)(44-46). In vivo studies supported the potent activity of zuranolone to modulate GABA-A receptors in the central nervous system after oral dosing in rats and mice(44-46). Allopregnanolone analogs protected mice from chronic stress-induced dysfunctions of neural network and behavioral states, and correlated with modulation of q oscillations in the basolateral amygdala, an important structure involved in mood regulation(44-46). An inhibitory effect of brexanolone on the inflammatory signaling post-infusion has been suggested in patients with PPD(47).
Brexanolone, a soluble intravenous preparation of synthetic allopregnanolone, was investigated in one phase II and two phase III trials focused on the efficacy and safety of this antidepressant(40,48,49). Brexanolone is administered in patients with PPD in order to produce a stable serum level of allopregnanolone similar to third-trimester concentrations of pregnancy(49). The HAMD scores were significantly reduced by brexanolone versus placebo in patients with PPD, after a single 14-day course(40). A significant reduction of HAMD scores in patients with MDD at 15 and 28 days versus placebo was reported after brexanolone administration(40). Also, in patients diagnosed with bipolar disorder, the decrease in HAMD scores was important(40). The main adverse events were sedation, dizziness and headache(40). Unfortunately, brexanolone requires a long infusion time, hospitalizing the patient during treatment, and a risk evaluation and mitigation strategy program(40,50). The last aspect is a recommendation formulated by the FDA after 4.3% of the patients treated with brexanolone presented a loss or altered state of consciousness(50).
Brexanolone is administered i.v. during 60 hours, with an initial dose of 30 µg/kg/h (0-4 h), increased to 60 µg/kg/h (4-24 h), then to 90 µg/kg/h (24-52 h), or preserving the 60 µg dose for those who cannot tolerate higher doses; after 52 hours, the dose should be decreased to 60 µg/kg/h and after 56 hours and up to 60 hours, the dose should be decreased to 30 µg/kg/h (Figure 3)(51). The terminal half-life of brexanolone is 9 hours, its metabolism is non-CYP based, and the excretion is through feces and urine (47% and 42%, respectively)(51). There were no significant variations in the drug’s pharmacokinetics in patients with severe renal impairment or hepatic dysfunction(51).
Zuranolone (50 mg/day) was assessed in a double-blind phase III trial that enrolled 196 patients with peripartum depression (PPD, defined as an MDD episode during the third trimester of pregnancy or ≤4 weeks after the child’s birth) monitored for 45 days(52). Zuranolone was superior to placebo in decreasing the severity of depressive symptoms after three days (the study’s primary outcome), and the most common adverse events were somnolence, dizziness and sedation(31). Another large, phase III trial randomized 543 patients with MDD on zuranolone (50 mg/day) or placebo, and the treatment was maintained for 14 days; statistically significant improvement in depressive symptoms was reported at day 15, based on HAMD scores(41). This trial also reported greater improvements versus placebo starting rapidly, from the third day of the treatment(41). Zuranolone was well tolerated, with no significant safety findings(41).
Zuranolone is also explored as a treatment in patients with bipolar depression(44).
Zuranolone is administered in doses of 50 mg/day for 14 days, and the dosage can be reduced to 40 mg/day if a central nervous system (CNS) depressant effect occurs(53). Zuranolone can be used alone or as an adjunct to oral antidepressant therapy(53). In patients with severe hepatic/renal impairment, the daily dose recommended is 30 mg orally, for 14 days(53). The terminal half-life of zuranolone is 19.7-24.6 h in an adult population; this drug is metabolized through CYP3A4 isoenzymes; its excretion is done by the kidney (45%) and feces (42%)(53).
According to a recent meta-analysis (n=9 reports), the efficacy of zuranolone and brexanolone was supported by the percentage of patients with PPD who achieved therapeutic response and remission versus placebo, according to the HAMD score evolution(54). Also, the percentage of patients with MDD achieving HAMD response and remission was significantly increased during zuranolone versus placebo, using HAMD scores as the main outcome(54).
A 3b-methylated synthetic analog of allopregnanolone, named ganaxolone, is undergoing testing for severe PPD as an add-on to the ongoing antidepressant(48,55). Other products from the same category that are undergoing investigation for a large range of psychiatric and neurological disorders are an allopregnanolone prodrug (LYT-300), the a2/3-selective GABAkines (KRM-II-81), and the a2/3/5-preferring GABAkines (PF-06372865, darigabat)(56). This interest in agents that regulate the concentration of allopregnanolone derives from the fact that the concentration of this substance is decreased in pathologies like Alzheimer’s disease, Parkinson’s disease, posttraumatic stress disorder, anxiety disorders, multiple sclerosis etc.(57)
The intranasal administration of esketamine was approved by the FDA in March 2019 and by EMA in February 2021 to manage treatment-resistant major depression, as an add-on agent to orally administered SSRIs or serotonin and norepinephrine reuptake inhibitors (SNRIs)(58-60). Esketamine is intended as an acute short-term treatment of psychiatric emergencies from the spectrum of depressive disorders that have been nonresponsive to first-line antidepressants, in order to obtain a rapid reduction of mood symptoms(58).
Esketamine’s mechanism of action is very complex and involves specific blockade of glutamate NMDA receptors containing GluN2B subunits, an NMDA receptor inhibition independent effect, and also the ability to induce a dopamine release in the striatum and other areas involved in the reward system(61).
In two phase III studies, TRANSFORM-1 (N=346 patients with moderate/severe MDD, non-responsive to at least two antidepressants) and TRANSFORM-3 (N=138 patients with treatment-resistant depression), esketamine added to oral antidepressants was not superior to placebo added to oral antidepressants for the primary endpoint (MADRS score)(62,63). Another phase III trial, TRANSFORM-2 (N=227 patients with treatment-resistant MDD), supported a significant superior efficacy of esketamine + antidepressant in reducing the MADRS scores versus placebo + antidepressant(64). Other two phase III trials, SUSTAIN-1 and 2, that enrolled 297 and 802 patients, respectively, assessed the long-term effects of esketamine administration as an add-on to oral antidepressants: the risk of relapse after remission/response under esketamine plus antidepressant was lowered by 51-70% during the 16 weeks of monitoring if the treatment continued, in the first trial. In the second trial, the duration of monitoring was 48 weeks, and the improvement of clinical status reached by the use of esketamine plus antidepressant was maintained with decreased MADRS scores(65,66). A meta-analysis of five studies (N=774 patients with treatment-resistant major depression) showed that adjunctive esketamine was significantly more effective than placebo for MADRS score change, remission and therapeutic response (p<0.0001)(67).
Warnings included in the Summary of Product Characteristics of esketamine nasal spray state that, before esketamine initiation, blood pressure should be assessed, and a high value of this parameter indicates the need for careful risks/benefits weighing; if significant or unstable cardiovascular or respiratory diseases are present, additional precautions are required. After esketamine dosing, blood pressure should be reassessed approximately 40 minutes and subsequently; the risk of sedation, dissociation and high blood pressure requires monitoring by a healthcare professional (Figure 4)(58).

The induction phase of esketamine nasal spray includes 1-4 weeks, with a starting dose of 56 mg, with subsequent doses of 56-84 mg twice a week; the maintenance phase includes weeks 5-8 with 56-84 mg once weekly doses, and from week 9 onwards, the administration of 56-84 mg every two weeks or once weekly(58). After the initiation phase, evidence of therapeutic benefit should be evaluated in order to assess the need for treatment continuation(58). In older patients (≥65 years old), the dose should be decreased to half, and the steps of the dose increments should be limited to 28 mg during the induction and the maintenance phase(58).
The absorption of esketamine is rapid through the nasal mucosa, with a Tmax=20-40 min, a distribution volume of 709 l (if administered i.v.), a 43-45% protein binding, and extensive metabolization in the liver (CYP2B6 and CYP3A4, but also CYP2C19 and CYP2C9)(58). Esketamine is eliminated by kidneys (approximately 78%) and by feces (approximately 2%)(58,61).
Toludesvenlafaxine was approved for MDD treatment by the Chinese National Medical Products Administration (NMPA) for clinical use in November 2022, and it has also been under review by the FDA since 2020(68,69).
This drug is the first-in-class triple reuptake monoamine inhibitor (Figure 5). According to the results of manufacturer-conducted clinical studies, toludesvenlafaxine (ansofaxine, anshufaxine) can significantly reduce anxiety, retardation/fatigue, anhedonia and cognitive symptoms, while facilitating faster social recovery in patients with MDD(68). This drug has no significant impact on sexual functioning, body weight and lipid metabolism, with a good overall tolerability(68).

The available data on the efficacy and tolerability of toludesvenlafaxine are derived from three phase I, one phase II and one phase III trials, with short duration (up to eight weeks)(70). In the phase II trial, 260 adult patients with MDD were randomized on 40, 80, 120 or 160 mg/day doses of toludesvenlafaxine or placebo, and a significant decrease in the HAMD scores was reported at week 6(71). A relatively high rate of adverse events was reported in this trial (52% versus 38.8%)(71). In a phase III trial, 558 adults with MDD received toludesvenlafaxine 80 or 160 mg/day or placebo, for eight weeks, and at the endpoint, MADRS scores decreased significantly in the active drug-treated groups(72). Nausea, headache, vomiting and drowsiness were the most frequently reported adverse effects of toludesvenlafaxine, but their incidence was lower than in the previously mentioned trial (approximately 5%)(72).
Also, preclinical studies exploring the acute and long-term effects of toludesvenlafaxine showed changes in prolactin and testosterone levels, rapid penetration into the striatum, and an increase of all three monoamine levels more than with desvenlafaxine(73,74). The affinity for the serotonin transporter (SERT), norepinephrine transporter (NET), and dopamine transporter (DAT) was defined by IC50 values of 31.4±0.4, 586.7±83.6, and 733.3±103 nM in studies that utilized in vitro measurements(75).
Toludesvenlafaxine is rapidly absorbed and transformed into O-desvenlafaxine after oral administration, and high concentrations have been detected in the hypothalamus(75). Morphological studies revealed increased density of dendritic spines in hippocampal CA1 neurons after toludesvenlafaxine administration in a “two-hit” stress mouse model(76).
Conclusions
Six new pharmacological products with antidepressant properties have been approved by different national or regional medicines agencies in the last five years, which indicates an accelerated rhythm of research in this field. Many other investigational products for treating unipolar or bipolar depression are also in advanced phases of research, suggesting that promising perspectives exist for patients diagnosed with this type of pathology. Gepirone, the combination of bupropion and dextromethorphan, zuranolone, brexanolone, toludesvenlafaxine and esketamine are interesting therapeutic options that encompass pharmacological agents based on the traditional monoaminergic theory of depression, and agents with innovative mechanisms of action (glutamatergic modulation or allopregnanolone agonists). Although quite heterogenous from a pharmacodynamic perspective, this “new wave” of antidepressants arrived with a potential paradigm-shift promise: fewer adverse events (for gepirone and toludesvenlafaxine), faster therapeutic effect (zuranolone, brexanolone), increasing the response and remission rates in treatment-resistant MDD (for esketamine nasal spray), efficacy for PPD (brexanolone), pharmacodynamic complementarity with other antidepressants (zuranolone as add-on, esketamine nasal spray added to SNRI/SSRI), and multitarget properties (bupropion plus dextromethorphan).
Bibliografie
- Touloumis C. The burden and the challenge of treatment-resistant depression. Psychiatriki. 2021;32(Suppl.I):11-14.
- Keitner GI, Mansfield AK. Management of treatment-resistant depression. Psychiatr Clin North Am. 2012;35(1):249-65.
- Tranter R, O’Donovan C, Chandarana P, Kennedy S. Prevalence and outcome of partial remission in depression. J Psychiatry Neurosci. 2002;27(4):241-7.
- Vasiliu O. Effects of the selective serotonin reuptake inhibitors over coagulation in patients with depressive disorders – a systematic review and retrospective analysis. RJMM. 2019;CXXII(2):7-11.
- Vasiliu O. Investigational drugs for the treatment of depression (Part 2): Glutamatergic, cholinergic, sestrin modulators, and other agents. Front. Pharmacol. 2022;13:884155.
- Vasiliu O, Vasile D, Mangalagiu AG, Petrescu MB, Candea CA, Tudor C, Ungureanu D, et al. Current treatment of posttraumatic stress disorder – A review of therapeutic guidelines and good practice recommendations. RJMM. 2020;CXXIII(4):241-248.
- Dubovsky SL. What is new about new antidepressants? Psychother Psychosom. 2018;87(3):129-139.
- Krystal JH, Charney DS., Duman RS. A new rapid-acting antidepressant. Cell. 2020;181(1):7.
- Jenkins SW, Robinson DS, Fabre LF Jr, Andary JJ, Messina ME, Reich LA. Gepirone in the treatment of major depression. J Clin Psychopharmacol. 1990;10(3 Suppl):77S-85S.
- Rickels K, Schweizer E, DeMartinis N, Mandos L, Mercer C. Gepirone and diazepam in generalized anxiety disorder: a placebo-controlled trial. J Clin Psychopharmacol. 1997;17(4):272-7.
- Leslie RA. Gepirone. Organon. Curr Opin Investig Drugs. 2001;2(8):1120-7.
- Yocca FD. Neurochemistry and neurophysiology of buspirone and gepirone: interactions at presynaptic and postsynaptic 5-HT1A receptors. J Clin Psychopharmacol. 1990;10(3 Suppl):6S-12S.
- BioSpace. Fabre-Kramer Pharmaceuticals announces FDA approval of EXXUATM, the first and only oral selective 5HT1A receptor agonist for the treatment of major depressive disorder in adults. https://www.biospace.com/article/releases/fabre-kramer-pharmaceuticals-announces-fda-approval-of-exxua-the-first-and-only-oral-selective-5ht1a-receptor-agonist-for-the-treatment-of-major-depressive-disorder-in-adults (accessed 16 October 2023).
- Gallagher A. FDA approves gepirone extended-release for treatment of major depressive disorder. Pharmacy Time. https://www.pharmacytimes.com/view/fda-approves-gepirone-extended-release-for-treatment-of-major-depressive-disorder (accessed 16 October 2023).
- University of Pennsylvania. Gepirone vs. placebo in the treatment of cocaine dependence-3. NCT00000189. https://clinicaltrials.gov/study/NCT00000189 (accessed 16 October 2023).
- Csanalosi I, Schweizer E, Case WG, Rickels K. Gepirone in anxiety: a pilot study. J Clin Psychopharmacol. 1987;7(1):31-3.
- Feiger AD, Heiser JF, Shrivastava RK, Weiss KJ, Smith WT, Sitsen JMA, Gibertini M. Gepirone extended-release: new evidence for efficacy in the treatment of major depressive disorder. J Clin Psychiatry. 2003;64(3):243-9.
- Fabre LF, Clayton AH, Smith LC, Goldstein I, Derogatis LR. The effect of gepirone-ER in the treatment of sexual dysfunction in depressed men. J Sex Med. 2012;9(3):821-9.
- Fabre LF, Smith LC, DeRogatis LR. Gepirone-ER treatment of low sexual desire associated with depression in women as measured by the DeRogatis Inventory of Sexual Function (DISF) fantasy/cognition (desire) domain - a post hoc analysis. J Sex Med. 2011;8(9):2569-81.
- Fabre LF, Brown CS, Smith LC, Derogatis LR. Gepirone-ER treatment of hypoactive sexual desire disorder (HSDD) associated with depression in women. J Sex Med. 2011;8(5):1411-9.
- Robinson DS, Sitsen JM Ad, Gibertini M. A review of the efficacy and tolerability of immediate-release and extended-release formulations of gepirone. Clin Ther. 2003;25(6):1618-33.
- Exxua®. Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/021164s000lbl.pdf (accessed 16 October 2023).
- Kaur GA, Bansal Y, Bhandari R, Kaur S, Kaur J, Singh R, et al. Gepirone hydrochloride: a novel antidepressant with 5HT1A agonistic properties. Drugs of Today. 2019;55(7):423.
- Auvelity®. Highlights of prescribing information. ttps://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215430s000lblCorrect3.pdf (accessed 16 October 2023).
- Keam SJ. Dextromethorphan/Bupropion: First approval. CNS Drugs. 2022;36(11):1229-1238.
- Stahl SM. Dextromethorphan/bupropion: A novel oral NMDA (N-methyl-D-aspartate) receptor antagonist with multimodal activity. CNS Spectr. 2019;24(5):461-466.
- Iosifescu DV, Jones A, O’Gorman C, Streicher C, Feliz S, Fava M, Tabuteau H. Efficacy and safety of AXS-05 (dextromethorphan-bupropion) in patients with major depressive disorder: A phase 3 randomized clinical trial (GEMINI). J Clin Psychiatry. 2022;83(4):21m14345.
- Tabuteau H, Jones A, Anderson A, Jacobson M, Iosifescu DV. Effect of AXS-05 (dextromethorphan-bupropion) in major depressive disorder: A randomized double-blind controlled trial. Am J Psychiatry. 2022;179(9):490-499.
- BioSpace. Axsome Therapeutics announces topline results of the STRIDE-1 phase 3 trial in treatment resistant depression and expert call to discuss clinical implications. https://www.biospace.com/article/releases/axsome-therapeutics-announces-topline-results-of-the-stride-1-phase-3-trial-in-treatment-resistant-depression-and-expert-call-to-discuss-clinical-implications/ (accessed 17 October 2023).
- European Medicines Agency. Nuedexta. https://www.ema.europa.eu/en/medicines/human/EPAR/nuedexta (accessed 16 October 2023).
- Kelly TF, Lieberman DZ. The utility of the combination of dextromethorphan and quinidine in the treatment of bipolar II and bipolar NOS. J Affect Disord. 2014;167:333-5.
- Schatzberg AF. Understanding the efficacy and mechanism of action of a dextromethorphan-bupropion combination: Where does it fit in the NMDA versus mu-opioid story? Am J Psychiatry. 2022;179(9):448-450.
- Nicholson KL, Hayes BA, Balster RL. Evaluation of the reinforcing properties and phencyclidine-like discriminative stimulus effects of dextromethorphan and dextrorphan in rats and rhesus monkeys. Psychopharmacology (Berl.) 1999;146:49-59.
- Akbar D, Rhee TG, Ceban F, Ho R, Teopiz KM, Cao B, et al. Dextromethorphan-bupropion for the treatment of depression: A systematic review of efficacy and safety in clinical trials. CNS Drugs. 2023;37(10):867-881.
- Pedraz-Petrozzi B, Deuschle M, Gilles M. Improvement of depressive symptoms, after a suicide attempt, with dextromethorphan/bupropion combination treatment in a patient with treatment-resistant depression and psychiatric comorbidities. Clin Case Rep. 2023;11(3):e7045.
- Majeed A, Xiong J, Teopiz KM, Ng J, Ho R, Rosenblat JD, et al. Efficacy of dextromethorphan for the treatment of depression: a systematic review of preclinical and clinical trials. Expert Opin Emerg Drugs. 2021;26(1):63-74.
- Parincu Z, Iosifescu DV. Combinations of dextromethorphan for the treatment of mood disorders – a review of the evidence. Expert Rev Neurother. 2023;23(3):205-212.
- Murrough JW, Wade E, Sayed S, Ahle G, Kiraly DD, Welch A, et al. Dextromethorphan/quinidine pharmacotherapy in patients with treatment resistant depression: A proof of concept clinical trial. J Affect Disord. 2017;218:277-283.
- Pine DS. Zuranolone treatment for depression: Steady progress in mechanism-focused therapeutic? Am J Psychiatry. 2023;180(9):631-633.
- Walkery A, Leader LD, Cooke E, VandenBerg A. Review of allopregnanolone agonist therapy for the treatment of depressive disorders. Drug Des Devel Ther. 2021;15:3017-3026.
- Clayton AH, Lasser R, Parikh SV, Iosifescu DV, Jung JA, Kotecha M, et al. Zuranolone for the treatment of adults with major depressive disorder: A randomized, placebo-controlled phase 3 trial. Am J Psychiatry. 2023;180(9):676-684.
- US FDA. FDA approved first treatment for post-partum depression. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-post-partum-depression (accessed 18 October 2023).
- US FDA. FDA approves first oral treatment for postpartum depression. https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-treatment-postpartum-depression (accessed 18 October 2023).
- Althaus AL, Ackley MA, Belfort GM, Gee SM, Dai J, Nguyen DP, et al. Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator. Neuropharmacology. 2020;181:108333.
- Antonoudiou P, Colmers PLW, Walton NL, Weiss GL, Smith AC, Nguyen DP, et al. Allopregnanolone mediates affective switching through modulation of oscillatory states in the basolateral amygdala. Biol Psychiatry. 2022;91(3):283-293.
- Zorumski CF, Mennerick S. Neurosteroids as therapeutic leads in psychiatry. JAMA Psychiatry. 2013;70(7):659-660.
- Patterson R, Balan I, Morrow AL, Meltzer-Brody S. Novel neurosteroid therapeutics for post-partum depression: perspectives on clinical trials, program development, active research, and future directions. Neuropsychopharmacology. 2023;10.1038/s41386-023-01721-1.
- Frieder A, Fersh M, Hainline R, Deligiannidis KM. Pharmacotherapy of postpartum depression: Current approaches and novel drug development. CNS Drugs. 2019;33(3):265-282.
- Faden J, Citrome L. Intravenous brexanolone for postpartum depression: what it is, how well does it work, and will it be used? Ther Adv Psychopharmacol. 2020;10:2045125320968658.
- Kleinman RA, Schatzberg AF. Understanding the clinical effects and mechanisms of action of neurosteroids. Am J Psychiatry. 2021;178(3):221-223.
- Zulresso®. Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211371lbl.pdf (accessed 18 October 2023).
- Deligiannidis KM, Meltzer-Brody S, Maximos B, Peeper EQ, Freeman M, Lasser R, et al. Zuranolone for the treatment of postpartum depression. Am J Psychiatry. 2023;180(9):668-675.
- Zurzuvae®. Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217369s000lbl.pdf (accessed 18 October 2023).
- Zou J, Yang L, Yang G, Gao J. The efficacy and safety of some new GABAkines for treatment of depression: A systematic review and meta-analysis from randomized controlled trials. Psychiatry Res. 2023;328:115450.
- Dichtel LE, Nyer M, dording C, Fisher LB, Cusin C, Shapero BG, et al. Effects of open-label, adjunctive ganaxolone on persistent depression despite adequate antidepressant treatment in postmenopausal women: A pilot study. J Clin Psychiatry. 2020;81(4):19m12887.
- Cerne R, Lippa A, Poe MM, Smith JL, Jin X, Ping X, et al. GABAkines – Advances in the discovery, development, and commercialization of positive allosteric modulators of GABAA receptors. Pharmacol Ther. 2022;234:108035.
- Bhatti NA, Jobilal A, Asif K, Villegas MJ, Pandey P, Tahir AN, et al. Exploring novel therapeutic approaches for depressive disorders: The role of allopregnanolone agents. Cureus. 2023;15(8):e44038.
- Spravato®. Summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/spravato-epar-product-information_en.pdf (accessed 18 October 2023).
- Johnson&Johnson. Spravato® (Esketamine nasal spray) authorised in Europe for the rapid reduction of depressive symptoms in a psychiatric emergency for patients with major depressive disorder. https://www.jnj.com/spravato-esketamine-nasal-spray-authorised-in-europe-for-the-rapid-reduction-of-depressive-symptoms-in-a-psychiatric-emergency-for-patients-with-major-depressive-disorder (accessed 18 October 2023).
- US FDA. FDA approves new nasal spray medication for treatment-resistant depression; available only at a certified doctor’s office or clinic. https://www.fda.gov/news-events/press-announcements/fda-approves-new-nasal-spray-medication-treatment-resistant-depression-available-only-certified (accessed 18 October 2023).
- Vasiliu O. Esketamine for treatment-resistant depression: A review of clinical evidence. Experim Ther Med. 2023;25:111.
- Fedgchin M, Trivedi M, Daly EJ, Melkote R, Lane R, Lim P, et al. Efficacy and safety of fixed‑dose esketamine nasal spray combined with a new oral antidepressant in treatment‑resistant depression: Results of a randomized, double‑blind, active‑controlled study (TRANSFORM‑1). Int J Neuropsychopharmacol. 2019;22(10):616‑630.
- Ochs‑Ross R, Daly EJ, Zhang Y, Lane R, Lim P, Morrison RL, et al. Efficacy and safety of esketamine nasal spray plus an oral antidepressant in elderly patients with treatment‑resistant depression – TRANSFORM 3. Am J Geriatr Psychiatry. 2020;28(2):121‑141.
- Popova V, Daly EJ, Trivedi M, Cooper K, Lane R, Lim P, et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment‑resistant depression: A randomized double‑blind active‑controlled study. Am J Psychiatry. 2019;176(6):428‑438.
- Daly EJ, Trivedi MH, Janik A, Li H, Zhang Y, Li X, Lane R, et al. Efficacy of esketamine nasal spray plus oral antidepressant treatment for relapse prevention in patients with treatment‑resistant depression: A randomized clinical trial. JAMA Psychiatry. 2019;76(9):893‑903.
- Wajs E, Aluisio L, Holder R, Daly EJ, Lane R, Lim R, et al. Esketamine nasal spray plus oral antidepressant in patients with treatment‑resistant depression: Assessment of long‑term safety in phase 3, open‑label study (SUSTAIN‑2). J Clin Psychiatry. 2020;81(3):19m12891.
- Papakostas GI, Salloum NC, Hock RS, Jha MK, Murrough JW, Mathew SJ, et al. Efficacy of esketamine augmentation in major depressive disorder: A meta-analysis. J Clin Psychiatry. 2020;81(4):19r12889.
- Luye Pharma. Luye Pharma’s Class 1 innovative antidepressant Ruoxinlin® approved for launch in China. https://www.luye.cn/lvye_en/view.php?id=2108 (accessed 18 October 2023).
- Luye Pharma. NDA filing for Luye Pharma’s antidepressant drug LY03005 accepted by US FDA. Retrieved online: https://www.luye.cn/lvye_en/view.php?id=1809 (accessed 16 October 2023).
- Vasiliu O. Efficacy, tolerability, and safety of toludesvenlafaxine for the treatment of major depressive disorder – A narrative review. Pharmaceuticals. 2023;16(3):411.
- Luye Pharma. Luye Pharma’s class 1 new drug anshufaxine hydrochloride extended-release tablets meets predefined endpoints in phase III trial. https://www.luye.cn/lvye_en/view.php?id=1922 (accessed 18 October 2023).
- Mi W, Yang F, Li H, Xu X, Li L, Tan Q, et al. Efficacy, safety, and toludesvenlafaxine of ansofaxine (LY03005) extended-release tablet for major depressive disorder: A randomized, double-blind, placebo-controlled, dose-finding, phase 2 clinical trial. Int J Neuropsychopharmacol. 2021;25:252-260.
- Li C, Jiang W, Gao Y, Lin F, Zhu H, Wang H, et al. Acute, subchronic oral toxicity, and genotoxicity evaluations of LPM570065, a new potent triple reuptake inhibitor. Regul Toxicol Pharmacol. 2018;98:129-139.
- Zhang R, Li X, Shi Y, Shao Y, Sun K, Wang A, et al. The Effects of LPM570065, a Novel Triple Reuptake Inhibitor, on Extracellular Serotonin, Dopamine and Norepinephrine Levels in Rats. PLoS One. 2014;9:e91775.
- Zhu H, Wang W, Sha C, Guo W, Li C, Zhao F, et al. Pharmacological Characterization of Toludesvenlafaxine as a Triple Reuptake Inhibitor. Front Pharmacol. 2021;12:741794.
- Meng P, Li C, Duan S, et al. Epigenetic Mechanism of 5-HT/NE/DA Triple Reuptake Inhibitor on Adult Depression Susceptibility in Early Stress Mice. Front Pharmacol. 2022;13:848251.