Electromyography is a method of recording electrical muscle activity during reflex or voluntary contractions. It offers information about neuromuscular physiology, being useful in highlighting afflicted structures in various neurological diseases, and especially in the diagnosis of muscle and neuromuscular diseases. In children, the importance of using cutaneous electromyography is given by the fact that it allows the diagnosis of progressive muscular dystrophies (Duchenne, the benign Becker-Kiener form), of congenital myopathies, as well as myositis of various causes (viral infections, inflammatory diseases, drugs or intense physical exercise).
REVIEW
Importanţa electromiografiei la copii
Importance of electromyography in children
First published: 30 decembrie 2023
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Pedi.68.4.2022.7523
Abstract
Rezumat
Electromiografia este o metodă de înregistrare a activităţii musculare electrice în timpul contracţiilor reflexe sau voluntare, oferind informaţii despre fiziologia neuromusculară, care sunt utile în evidenţierea structurilor afectate în diverse afecţiuni neurologice şi mai ales în diagnosticarea afecţiunilor musculare şi neuromusculare. La copii, importanţa utilizării electromiografiei cutanate este dată de faptul că permite diagnosticarea distrofiilor musculare progresive (Duchenne, forma benignă Becker-Kiener), a miopatiilor congenitale, precum şi a miozitei de diverse cauze (infecţii virale, boli inflamatorii, medicamente sau exerciţii fizice intense).
Introduction
Electromyography is a method of recording electrical muscle activity during reflex or voluntary contractions. Electromyogram is the graphic recording of the biopotentials generated by muscle activity. The principle of electromyography is based on the propagation of electrical currents generated by muscle action potentials at a distance from the site of production, thus allowing them to be detected and collected by a special device called electromyograph.
Electromyography is thus a method of investigation of neuromuscular physiology, useful in highlighting affected structures in various neurological diseases, and especially in the diagnosis of muscle and neuromuscular diseases.
Both surface and needle electrodes can be used to collect muscle action biopotentials. Surface electrodes are placed on the tegument above the muscle being examined, and provide information on the overall electrical activity of the muscle. Because they represent a noninvasive method, these electrodes are used to perform skin electromyography in pediatric practice. Needle electrodes allow both the investigation of small portions of muscle and the study of motor unit potentials(30). The axons of the spinal cord motoneurons innervate skeletal muscle fibers. A single motor neuron is responsible for innervating about 600 muscle fibers, whereas in a human body there are about 250 million muscle fibers and only 420,000 motor neurons.
The action potential invades the terminal bouton, producing the release of acetylcholine at the neuromotor plate. This diffuses through the synaptic cleft and acts on nicotinic receptors, leading to depolarization of muscle fibers and, ultimately, to muscle contraction. This depolarization electric field is recorded by electrodes placed on the skin(23).
Skin electromyography thus assesses muscle function by recording muscle activity on the surface of the integument, with the disadvantage of providing a limited assessment of muscle activity(10). It is, however, preferable in pediatric practice, due to the noninvasive nature of the method. Skin electromyography can be recorded by a pair of electrodes or by a complex set of multiple electrodes, as electromyographic recordings display the potential difference between at least two different electrodes. Other disadvantages of this method are also related to the fact that skin electrode recordings are limited to superficial muscles and are influenced by the depth of subcutaneous cellular tissue, which can vary depending on the patient’s weight(25).
In terms of clinical examination, electromyographic recording plays a key role in the assessment of neuromuscular disorders in infants and children, as there are a large number of such disorders in the pediatric age group. In some of these cases, electromyography establishes a diagnosis, and in many other situations it is used to guide further evaluation – for example, genetic testing or muscle biopsy(35).
Electromyograph
Electromyograph is the equipment used to detect and record electrical activity of muscles under resting and contracted conditions, as well as under normal or pathological conditions(17).
It consists of:
1. Harvesting electrodes – take the form of silver pads of varying sizes, which are applied to the tegument over the muscle to be explored, usually at the proximal and distal ends of the muscle, so that the axis intersecting the two electrodes is parallel to the muscle fibers.
2. Amplification system – its role is to amplify the biocurrents produced, being particularly useful in situations where spontaneously occurring biocurrents have a very low amplitude (100 µV).
3. Display system – this can be a monitor, resulting in a digital display of the route, or it can be in the classic paper format(23).
Examination technique
Before an electromyography is performed, certain conditions must be met in order to increase patient’s comfort, especially considering that the patient is of pediatric age. First of all, the room in which the investigation is carried out should have an ambient temperature of around 22-24ºC, with the proviso that, at lower temperatures, in addition to the discomfort of the child, the pathway may change due to involuntary muscle contractions produced as part of the thermoregulation phenomenon(23).
The patient should be explained in an age-appropriate way what the examination is about, in order to remove any anxiety or fear. It is important for the doctor to have a friendly attitude and gain the patient’s trust, as some functional parameters can also be influenced to some extent by the patient’s mental state. The examination should also be carried out in a position that is comfortable for the patient, preferably sitting or lying on a bed, without forced attitudes, which allows good muscle relaxation.
After the patient is given the necessary explanations, he is invited to sit comfortably and relaxed on the examination bed, and during this time the doctor establishes the examination plan and the order in which the muscles will be examined. Next, the electrodes are placed on the skin, and the examiner watches the display system to see whether or not electrical activity is occurring, asking the patient to perform a voluntary muscle contraction(20).
Normal electromyography. Physiological variations
At rest, the electromyograph does not detect any bioelectrical activity, so an isoelectric trace is shown on the display. A mild, voluntary muscle contraction causes a simple electromyographic trace to appear, consisting of monophasic or biphasic action potentials. As the force of contraction increases, more waves appear on the pathway due to spatial and temporal summation phenomena. When the muscle is fully contracted, a disordered group of action potentials with different durations and amplitudes should appear(30).
An electromyographic tracing may show changes of no pathological significance, caused by a number of physiological factors. This is important to be aware of because physiological changes need to be taken into account when interpreting the results.
An example of such a factor is age, a change that is all the more important as this paper refers to electromyographic tracings at pediatric age(26). It is thus considered that the duration and amplitude of action potentials are directly proportional to age, this being due to the fact that neuromuscular synapses in children have a smaller surface area. Further growth and development of the body also result in an increase in the volume of neuromuscular synapses(20).
Another factor that produces changes in the pathway is temperature, because the thermoregulation mechanism of the human body has a neuromuscular component and any thermal variation during the examination produces changes in the electromyographic pathway. It is therefore necessary to have a constant temperature of 22-24ºC in the examination room, at which temperature the thermoregulation mechanisms are not activated. In conditions of increasing body temperature (presence of an infection), no noticeable changes in the pathway occur, but in the case of hypothermia (common in the extremities) the changes are well known and characterized by a decrease in the amplitude of the pathway and a decrease in the ability to achieve a maximum contraction(35).
Like hypothermia, muscle fatigue affects neuromuscular transmission because the amplitude is lower in a tired muscle(10).
Diagnosis of neuromuscular disorders
Neuromuscular disorders differ in case of children and adults(8). Adults are more likely to have an electromyogram and the most common conditions diagnosed include radiculopathy, polyneuropathy and carpal tunnel syndrome. On the other hand, the neuromuscular disorders seen in children are quite different. For example, neuropathies are very common in adults, but extremely rare in children. Peripheral neuropathies that occur in children are most often genetic, whereas in adults they have a toxic, metabolic or inflammatory etiology. On the other hand, radiculopathies are only found in children in cases of trauma(18).
The most common electromyographic diagnoses in children are inherited motor neuron (spinal muscular atrophy), peripheral nerve (Charcot-Marie-Tooth disease) or muscle (muscular dystrophy) disorders(16).
Children with neuromuscular disorders often present as a clinical manifestation a motor delay(12). In most cases, it is not possible to determine from signs and symptoms alone whether the etiology is central or peripheral, and electromyographic examination is very useful in this respect as it allows the differentiation of central etiology from peripheral etiology, and the appropriate guidance for further assessment(24).
Pediatric neuromuscular pathology
1. Progressive muscular dystrophies
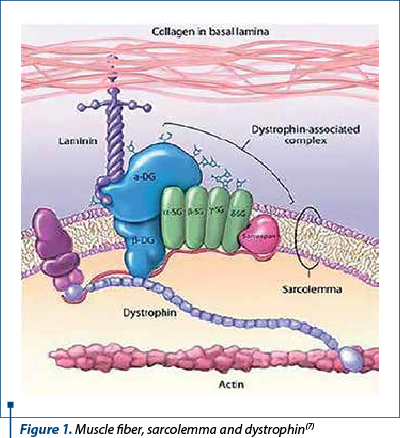
The term muscular dystrophy refers to a group of conditions involving progressive loss of muscle mass, resulting in muscle weakness and disability. The onset is insidious in the early years of life, with predominantly proximal muscle damage, abolition of osteotendinous reflexes and pseudohypertrophy of the muscles, particularly in the calves(32). The cause of dystrophy is a genetic mutation that interferes with the production of a cytoskeletal protein (dystrophin) that plays a role in membrane stabilization, therefore its deficiency results in disruption of the sarcolemma with excessive Ca2+ entry, resulting in prolonged muscle fiber contraction followed by necrosis (Figures 1 and 2). Lack of dystrophin causes cell instability and progressive loss of intracellular components, leading to increased creatine phosphokinase (CPK) levels(7).
Dystrophin is not only located in skeletal muscle, but is also found in smooth muscle and myocardium, as well as in the brain.
As the condition progresses, patients become increasingly immobile, and in some cases the respiratory muscles or myocardium may be affected, leading to serious complications. There is currently no method to prevent or treat muscular dystrophy, with the only existing therapies and drug treatments aimed at relieving symptoms, improving quality of life and slowing the progression of the disease(37).
Clinically, at least five major clinical forms of progressive muscular dystrophy can be described:
-
Duchenne muscular dystrophy
-
Becker muscular dystrophy
-
Emery-Dreifuss muscular dystrophy (humeroperoneal)
-
Fascioscapulohumeral muscular dystrophy (Landouzy-Dejerine)
-
Various dystrophies of the pelvic girdle.
The following will describe the first three clinical forms of muscular dystrophy, being the most common in current clinical practice.
1.1. Duchenne muscular dystrophy
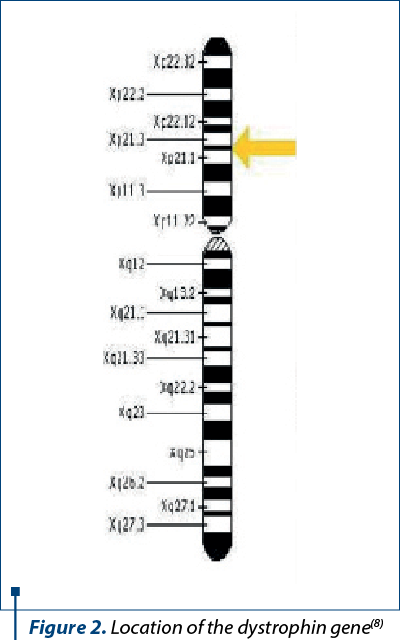
Duchenne muscular dystrophy (DMD) is the most common form of dystrophy, being an autosomal recessive inherited disorder linked to the X chromosome, characterized by a defect in the dystrophin gene located on the short arm of the X chromosome (Xp21.2). Thus, Duchenne muscular dystrophy affects only male newborns, with an incidence of about 1/3500 live male newborns. Girls carrying the gene responsible for the development of dystrophy are asymptomatic, and in rare cases they may show mild pseudohypertrophy and hypotonia of the pelvic muscles(9).
The condition begins around the age of 3-5 years old, with motor deficits and symmetrical amyotrophies, initially affecting the pelvifemoral, then the scapulohumeral girdle, with progressive extension to other muscle groups, with patients becoming wheelchair dependent around the age of 12(40).
The affected children differ from the start in that they require more time and effort to learn to walk, and later they have a difficult, swaying or tippy-toeing gait. The average age at which boys with Duchenne muscular dystrophy are able to walk is around 18 months old. They may also have difficulty running with a tendency to fall, climbing up and down stairs and getting up from the ground by climbing on their own body(4).
Cardiac damage is represented by dilated cardiomyopathy, following fibrosis, as well as rhythm and conduction disorders. Clinically manifest cardiomyopathy occurs after the age of 10 years old, affects one third of patients aged 14 years old, and is present in all patients over 18 years old. A persistent tachycardia, considered to be one of the preclinical manifestations, has been targeted in a proportion of patients under 6 years of age(31).
The respiratory function is impaired in all patients, who present with chronic restrictive respiratory failure due to intercostal muscle damage. Vital capacity is normal up to the age of 10 years old, and decreases after this age at a rate of 8-12% per year. When the vital capacity falls below 1 liter, the risk of death within two years is extremely high(5).
The intellectual development is delayed in some children, accompanied by brain atrophy and behavioral disorders, the severity of which is not correlated with the severity of hypotonia.
The digestive function is also affected by the occurrence of gastric hypomotility, and the patients may present episodes of abdominal pain, vomiting and abdominal distension with pseudo-obstructive clinical picture. These manifestations occur as a result of changes in the visceral smooth muscles.
The osteoarticular system is affected by the development of scoliosis, with a major impact on the respiratory function. As a result of frequent falls and bone demineralization, numerous fractures of the long bones occur(5).
The course of the disease is severe, with progressive worsening, so that death occurs around the age of 20 through numerous possible respiratory complications or heart failure(39).
Paraclinical investigations
-
Muscle enzymes are elevated at least 10-20 times the normal level, sometimes even in newborns and in the absence of any symptoms, even considered a screening method at birth. With advancing age, enzyme values begin to fall as a result of progressive muscle fiber destruction(38).
-
Electromyography allows the differential diagnosis with neuropathic muscular hypotonia, where there is spontaneous resting activity and action potentials with increased duration and amplitude. The electromyographic trace is myopathic, of short duration, with low amplitude action potentials(20).
-
Muscle biopsy is no longer considered a routine test, especially in the presence of positive genetic tests and symptoms. It is only performed in situations where other tests have been inconclusive or the genetic testing is negative(38).
Treatment
The treatment objectives are represented primarily by the prevention of osteoarticular deformities through physiotherapy and the use of special orthotics. Special attention is paid to wheelchair-bound patients, because in 90% of cases severe scoliosis occurs, which favors the aggravation of restrictive respiratory failure. Use torso supports, such as corsets or special braces(4).
In late stages with chronic respiratory failure, continuous positive pressure nasal ventilation is used. To prevent possible infections, pneumococcal and Haemophilus influenzae type B vaccine should be given to all patients.
If dilated cardiomyopathy is present, beta-blockers and angiotensin-converting enzyme inhibitors are recommended(31).
Corticosteroid therapy is the only therapeutic method that has shown some improvement. It is given to all patients, after informing them of possible side effects.
Myoblast transplantation or the replacement of dystrophin with a similar protein (utrophin) are other therapeutic methods, but they are still in their early stages(14).
1.2. Becker muscular dystrophy
Becker muscular dystrophy (BMD) is also a hereditary condition, with the same X-recessive inheritance, but with a slower progression and with milder clinical picture than that of Duchenne muscular dystrophy. This is explained by the fact that the stabilizing protein (dystrophin), which is absent in Duchenne dystrophy, is present in insufficient quantity and partially fulfils its function. The incidence in this case is 1/30,000 live male newborns(9).
The onset of the condition is variable, from 5 to 25 years old, with an average age of symptoms onset of 12 years old, and with the ability to walk preserved until the age of 16. Because patients do not become dependent on a wheelchair at an early age, spinal deformity is much rarer(5).
The clinical picture is similar to that of Duchenne dystrophy, but with less aggressive symptoms. Muscle weakness is manifested by difficulty walking, running and frequent falls. The pelvic girdle muscles are mainly affected, the Achilles reflexes are abolished, but pseudohypertrophy is very rarely present. Cardiac involvement (dilated cardiomyopathy with heart failure and rhythm disturbances) is rarer than in Duchenne muscular dystrophy, and when it occurs, it is not as severe. The intellectual function is not affected, but a range of concentration and interaction disorders may occur. Respiratory failure also sets in much more slowly, becoming symptomatic around the age of 40-50 years old(37).
Life expectancy is much higher, with most patients living to 60 years of age and death occurring from the same causes, recurrent lung infections and heart failure.
Paraclinical investigations
-
Muscle enzymes: significantly increased values.
-
Electromyography: again, it shows a myopathic pathway with low amplitude action potentials.
-
Muscle biopsy is useful for performing immunohistochemical investigations using monoclonal antibodies so that quantitative and/or qualitative dystrophin abnormalities can be revealed(40).
1.3. Emery-Dreifuss muscular dystrophy
Emery-Dreifuss muscular dystrophy is a rare hereditary disorder with autosomal dominant, recessive or X-linked inheritance, with an unknown overall incidence. X-linked transmitted disease affects 1/100,000 people, but women are carriers only, while men may be clinically affected. The incidence of the autosomal dominant transmitted type is unknown, but it is thought to be higher than the one of the X-linked form. The autosomal recessive type is the rarest, with only a few cases reported(21).
This condition is caused by a mutation in the gene coding for two membrane proteins: emerin and lamin A and C, which are essential for skeletal and cardiac muscle function. Emerin is a serine-rich membrane protein located on the inner face of the membrane of the nucleus of striated and myocardial muscle cells. Its main role is to help maintain the stability of nuclear structure and function, particularly in muscles that are highly stressed by contraction (increased calcium flux). Lamin A and C are also membrane proteins that interact with emerin and ensure that the nucleus is anchored to the plasma membrane and held inside the cell(9).
The clinical picture can vary from person to person, but symptoms most commonly begin between the ages of 5 and 15 years old, with Achilles tendon contractures, loss of joint mobility and stiffening of the cervical spine, muscle weakness in the peroneal muscles of the calves and in the muscles of the upper limbs, muscle atrophy, pathological tiptoeing and significant cardiomyopathy-like heart damage, rhythm and conduction disturbances, atrial paralysis, with an increased risk of sudden death(21).
The diagnosis is supported by the clinical picture, the patient’s age and the paraclinical investigations, where a slight increase in the level of muscle enzymes (CPK) is observed, but the abnormality is only confirmed by DNA tests.
The main objective of treatment is to prevent muscle strain and contractures and to use a pacemaker in patients with severe conduction disturbance (atrioventricular block)(41).
2. Inflammatory myopathies – dermatomyositis, polymyositis
Dermatomyositis is the most common idiopathic juvenile inflammatory myopathy, being a systemic vasculopathy affecting the skin and muscles, causing a progressive and symmetrical motor deficit of the girdle muscles, but remitting after immunosuppressive treatment(2).
The age of onset of the condition is 6-7 years old, with an incidence of 2-4 new cases/1 million children/year, mainly affecting females. The etiology is not fully elucidated, but a variety of infectious triggers (Coxsackie B virus, group A hemolytic Streptococcus b) are thought to act on a genetic predisposing terrain(3).
From an immunopathogenic point of view, following the onset of an immune response, capillary necrosis with perivascular inflammation, ischemia and subsequent destruction of muscle fibers gradually occur. Striated muscles thus lose their structure, showing areas of degeneration, interstitial edema and connective tissue proliferation. Excessive fibrosis leads to chronic inflammation(11).
In terms of clinical manifestations, the onset of the condition is insidious, often with low fever, asthenia and weight loss. The main damage is to the integument and muscles, but in some cases other organs and systems may also be affected: respiratory, cardiovascular, digestive, ocular etc.(27)
Cutaneous involvement is manifested by the appearance of erythema alternating with areas of atrophy, most commonly in areas directly exposed to the sun’s rays; subcutaneous edema causes thickening of the skin, and healing is achieved with remaining hyperpigmented lesions.
Muscle damage is important, being characterized by three major signs: myalgia, muscle swelling and muscle fatigue, occurring with small efforts. It is observed over time, as the child can no longer run as before, no longer climbs or descends stairs and lifting from ground level is done by climbing on his own body. Most of the striated muscle groups in the body are affected, including the extrinsic muscles of the eyeballs (causing diplopia), the flexor muscles of the neck (the child is unable to lift the head from a horizontal plane), the muscles of the back wall of the pharynx (dysphagia, dysphonia), but also the smooth muscles (in the gastrointestinal tract, abdominal pain, diarrhea and even lower gastrointestinal hemorrhages occur) and the muscles of the sphincters, causing incontinence. Manifestations such as muscle atrophy and tendon retractions may also be present.
Damage to other organs
-
respiratory: respiratory failure of the restrictive type, decreased vital capacity;
-
cardiac: myocarditis, dilated cardiomyopathy, rhythm and conduction disorders;
-
digestive: dysphagia, decreased intestinal absorption;
-
renal: proliferative glomerulonephritis;
-
ocular: retinitis, irritation;
-
joint manifestations caused by immobility;
-
central nervous system: seizures, depression(2,13).
Paraclinical investigations
Muscle enzymes: CPK, LDH, TGO and TGP have increased values, caused by chronic muscle inflammation.
Inflammatory anemia, VSH may be increased, lymphopenia with increased CD4/CD8 ratio.
Electromyography: a pathological pattern is recorded in 90% of cases; this may be of the myopathic type with fibrillatory, polyphasic potentials with high frequency and low amplitude, or with repetitive, high frequency burst action potentials. Electromyography is an indispensable method both for diagnosis and for subsequent monitoring of the evolution of the disease and the response to treatment; the comparative study of electromyographic patterns, observing the appearance or disappearance of bioelectrical abnormalities, can determine whether there has been an improvement or worsening of the pathological process(30).
Muscle biopsy reveals areas of muscle necrosis and perivascular inflammatory infiltrate as well as foci of regeneration.
Treatment
Its main objective is to improve the quality of life, increase muscle strength and relieve extramuscular manifestations.
Corticosteroids are administered according to severity. Thus, in forms with only cutaneous involvement, they are administered in low doses of 0.5 mg/kg b.w./day with monitoring of muscle enzymes; in forms with mild muscle damage, doses of 1-2 mg/kg b.w./day are administered, and in case of obtaining improvement, they are decreased to a maintenance dose of 5-15 mg/day. In severe forms of the disease, there are used pulses of methylprednisolone 30 mg/kg b.w./day, maximum 1 g/day, three consecutive days, then one pulse per week until the normalization of inflammatory markers.
Immunosuppression: if rapid response to corticosteroid therapy is not achieved, initiate therapy with methotrexate 15-20 mg/m2 in combination with folic acid 1 mg/day. In cases with unfavorable response to this type of therapy as well, cyclophosphamide 500 mg/m2 is given. The adverse effects of immunosuppression can be counteracted by the administration of immunoglobulin 0.4 g/kg b.w.
Therapies with anti-TNF-a biologic agents are also used (etanercept, infliximab) and anti-CD20 monoclonal antibodies (rituximab).
Other indicated therapies are physiotherapy, to prevent the onset of muscle contractions and atrophies; the dietary regime will be indicated for cases with dysphagia, by eating semi-liquid food or even installing a nasogastric feeding tube; for cases with severe respiratory failure, oxygen administration is recommended, and psychotherapy sessions are recommended for the child’s mental state.
The course of the disease can be favorable, about 50% of cases being cured with appropriate treatment, but a third remaining with major disabilities. It is important that the disease is diagnosed at an early stage, before muscle morphological changes become irreversible, in order to benefit from a favorable response to treatment. Without adequate treatment, or treatment initiated at a too advanced stage, the mortality rate is 40%, and the causes can include respiratory failure, arrhythmias, myocarditis, as well as hemorrhages and intestinal perforations(2,11,13).
Polymyositis is an inflammatory condition with systemic manifestations in which the main tissue affected is the muscle tissue, without skin involvement. Amyotrophies and muscle strength deficits reflect inflammatory, necrotic and degenerative histological changes. The disease can occur at any age, but is less common in children than dermatomyositis(2).
Muscle strength deficiency is the most common first symptom to appear, especially in the proximal muscles (waist and limbs), with motor difficulties in climbing up and down stairs, standing or lifting objects. Later, other muscle groups are also affected, such as the extrinsic muscles of the eyeballs, the muscles of the palate, the neck and the pharyngeal muscles. In acute cases of the disease, the muscles may become painful, swollen and indurated, with contractures leading to joint deformities. Injury to the pharyngeal and respiratory muscles can be fatal(29).
The diagnosis is established on the basis of laboratory changes, increased muscle enzymes and HSPV, electromyography which records a pathological pattern with fibrillation potentials and high-frequency discharges with sudden onset and termination, constant in amplitude, and on the basis of muscle biopsy which, again, shows areas of necrosis and inflammatory infiltrate.
3. Congenital myopathies
Congenital myopathies are a group of conditions characterized by histopathological changes in the muscle fiber, resulting in a disorganization of the internal architecture, unlike dystrophies, where the muscle fiber develops normally before degenerating. They occur in children from an early age, sometimes without progressive character, with a prevalence of 4-5 cases/10,000 children.
Most of these conditions manifest themselves immediately after birth by hypotonia, muscle weakness, abolished osteotendinous reflexes and delays in movement. Sometimes, the symptoms appear later in childhood or adolescence.
The diagnosis is established as in other muscle disorders, by determining the creatine kinase value, which may be normal or slightly increased, by electromyography showing a myopathic pathway with short action potentials, and by muscle biopsy to classify the condition. The biopsy most commonly reveals the predominance of type I muscle fibers or areas of hypotrophy, without fiber necrosis or other evidence of degeneration, which is important in the differential diagnosis with muscular dystrophies.
Nine clinicogenetic forms of congenital myopathies are described, depending on the location of the genetic defect, but the most common and best known forms are central core myopathy, non-malignant myopathy and centronuclear (myotubular) myopathy(36).
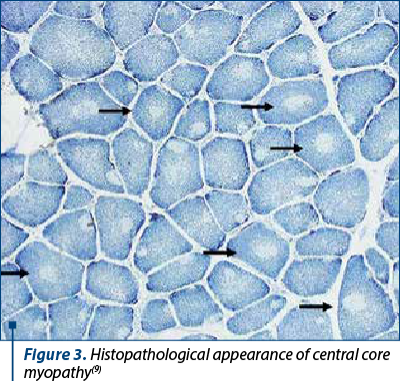
a) Central core myopathy is a congenital autosomal dominant myopathy, named so for the well-defined, round areas within the type I muscle fibers where there is no oxidative enzyme activity(22).
The symptoms are variable, and in addition to the hypotonia seen from birth, there are muscle weakness and delayed motor development. Children frequently develop skeletal malformations such as scoliosis and hip dislocation. This condition has a high risk of association with malignant hyperthermia syndrome when the patient undergoes surgery with general anesthesia, explained by the presence of a mutation in the RYR1 receptor genes(19).
The diagnosis is established both on the basis of symptoms and by investigations, which reveal normal creatine kinase values, a pathological electromyographic pattern with short-duration, low-amplitude action potentials, and is confirmed by muscle biopsy, which reveals a characteristic appearance with well-defined, rounded areas in the center of the muscle fiber (Figure 3).
There is no specific treatment for this condition, ventilatory support being recommended in cases with respiratory failure and the use of general anesthesia is avoided(22).
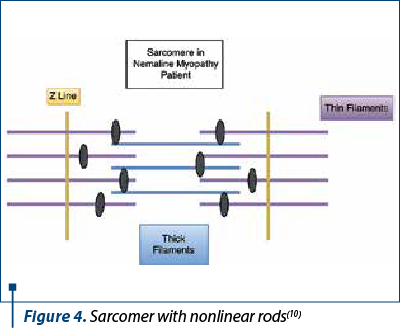
b) Nemaline myopathy is a congenital hereditary neuromuscular disorder with a prevalence of 1/50,000 live births. The main symptoms that can occur are muscle weakness, hypotonia, difficulty swallowing and impaired speech. The severe form may even manifest itself in the absence of breathing and spontaneous movements at birth, and may be associated with pathological contractures and fractures(34).
The contraction of muscle fibers occurs as a result of complex mechanical and chemical processes. When any part of the processes or fiber structure is disrupted, dysfunction results. In those with non-malignant myopathy, muscle contraction is impaired by the interposition of rod-like components in some muscle fibers (Figure 4). Their presence does not cause muscle weakness and there is no link between the number of rods and the degree of muscle weakness, they are only an indicator that there is a problem in the fiber. It is assumed that the arrangement of muscle fibers is affected, causing the muscles to be unable to contract effectively(1).
As with other myopathies, the diagnosis can be established by paraclinical investigations. Electromyography is the first examination performed and may reveal a pathological, myopathic pattern. Nuclear resonance imaging is also useful in this situation, as it provides an overview of the entire musculoskeletal system. Last but not least, muscle biopsy confirms the diagnosis by showing that the muscle fibers contain rod-like structures.
There is no specific treatment for this condition either, the only measures that can be applied are to relieve the symptoms. These measures vary depending on the severity of the disease, from moderate exercise to help keep muscles in good working order to the use of stabilizers such as braces. As children get older, it is essential for their development that they remain under the supervision of a multidisciplinary team of pediatrician, therapist, psychologist and speech therapist(36).
c) Centronuclear (myotubular) myopathy is a severe congenital myopathy characterized by abnormal localization of the muscle cell nucleus in the center of the muscle cell (Figure 5). It is considered the most serious myopathy because it is accompanied by neonatal hypotonia and severe, often fatal, respiratory failure(33).
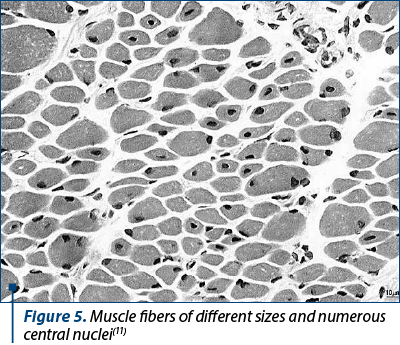
As with other myopathies, the clinical manifestations are mainly muscle weakness and associated disability. The child’s developmental milestones are delayed, particularly in motor acquisition, such as head control and gait. While some children reach adulthood with the ability to get around on their own, others become dependent on a wheelchair from an early age(36).
In order to establish the diagnosis, electromyographic examination is the first step, and it is also the one that objectifies the presence of a myopathy through abnormal spontaneous activity and low amplitude of action potentials. The disadvantage is that electromyography cannot specify what type of myopathy is present, so muscle biopsy and subsequent genetic testing are required.
There is no cure for centronuclear myopathies, but attempts are made to preserve functional abilities and decrease the risk of complications. The highest attention is paid to preventing lung infections, as these patients do not have the muscle strength to cough up secretions. Spinal alignment is also monitored, as weakening of the trunk muscles leads to scoliosis, which can compromise respiratory function. When it can no longer be prevented, surgery is performed to treat scoliosis(6).
Electromyographic traces
Normally, at rest the muscle is quietly electrical. This means that the electromyographic examination of the relaxed muscle does not reveal the existence of a bioelectrical activity, so that an isoelectrical line will appear on the screen of the device (Figure 6).
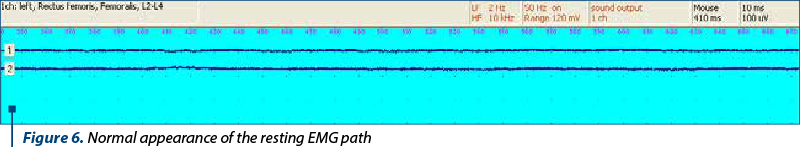
In children, under the conditions of examination, muscle rest (and, consequently, the electrical rest) must be obtained following a complete collaboration of the child, in order to achieve complete muscle relaxation.
Voluntary contraction
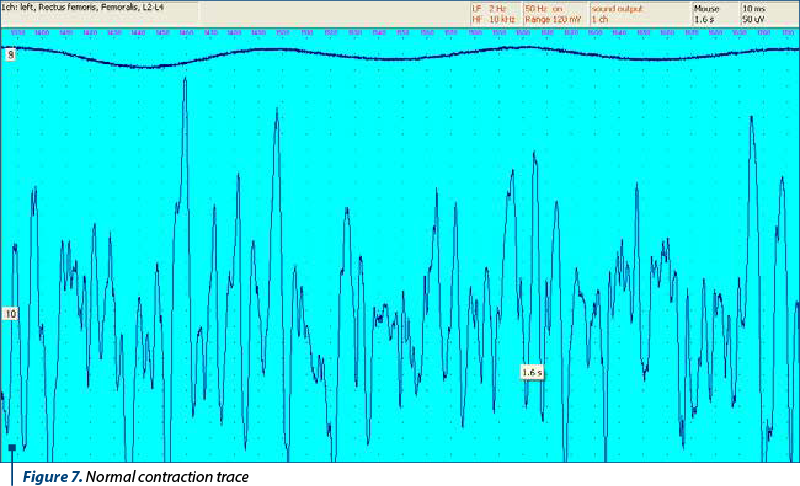
A mild muscle contraction causes the appearance of a simple electromyographic route, consisting of mono- or biphasic action potentials with an amplitude of 200-400 µV and a duration of 3-4 ms. As the force of contraction increases, the enrichment of the electromyographical path also occurs, due to the phenomena of temporal and spatial summation.
Damage to muscle fibers produces two types of pathological paths: electromyographic route of myogenous type and electromyographic route of neurogenic type.
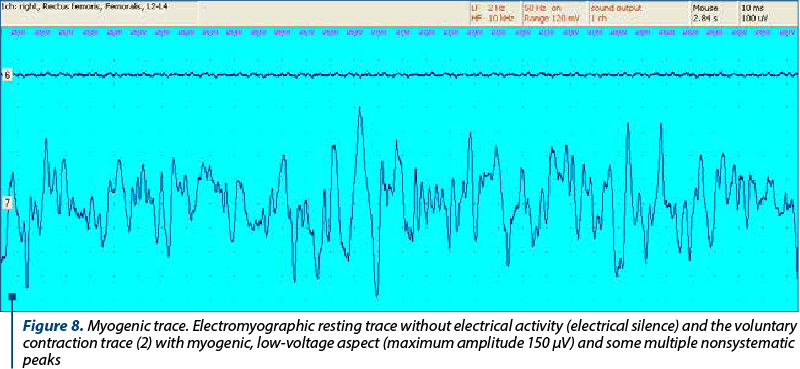
Features of the myogenic trace (Figure 8)
1. At rest: lack of spontaneous electrical activity (electrical silence).
2. In contraction: the path has over-added polyphasic potentials of short duration and low amplitude.
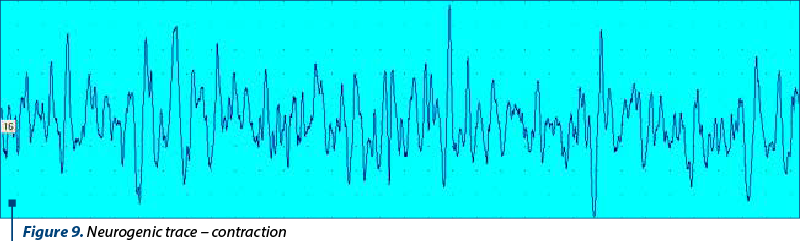
Features of the neurogenic trace (Figure 9)
1. At rest: spontaneous electrical activity manifested by potentials of fibrillation, fasciculation, denervation and re-innervation.
2. In contraction: route with its own rhythm of discharge, gradual contraction and the presence of late potentials after relaxation.
Conclusions
Neuromuscular symptoms are a frequent reason for presentation to the doctor in the pediatric population, being nonspecific for a particular pathology and requiring a thorough diagnostic approach. Any muscular or systemic condition with muscle involvement manifests itself in the child with hypotonia and myalgia, but the differential diagnosis is a complex one.
The surface electromyogram is a noninvasive investigation that revealed myogenous routes in most of the cases in the studied groups, which required further exploration. Electromyography in dynamics is a rapid complementary exploration that helps to differentiate between transient and chronic myopathies (congenital or acquired), until other diagnostic tests (genetic, biological) are available.
Conflict of interests: The author declares no conflict of interests.
Bibliografie
-
Agrawal P, Strickland C, Midgett C, et al. Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations, Ann Neurol. 2004;56(1):86-96.
-
Arion C. Polymyositis/Dermatomyositis. In: Ciofu EP, Ciofu C. (under ed.): Tratat de pediatrie, Bucharest, Ed. Medicală, 2001, 962-965.
-
Artlett CM, Ramos R, Jimines SA, et al. Chimeric cells of maternal origin in juvenile idiopathic inflammatory myopathies. Childhood Myositis Heterogeneity Collaborative Group. Lancet. 2000;356(9248):2155-2156.
-
Avramescu E. Duchenne muscular dystrophy. In: Kinetoterapia în afecţiuni pediatrice. Ed. Universitară, Craiova, 2007.
-
Bakker E, Van Ommen GJB. Duchenne and Becker Muscular Dystrophy (DMD and BMD). In: Neuromuscular Disorders: Clinical and Molecular Genetics, Edited by. A.E.H., Emery, John Wiley & Sons Ltd., 1998, 59 -85.
-
Biancalana V, Beggs AH, Das S, et al. Clinical utility gene card for: Centronuclear and myotubular myopathies. Eur J Hum Genet. 2012;20(10). doi:10.1038/ejhg.2012.91.
-
Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82(2):291-329. doi:10.1152/physrev.00028.2001.
-
Brooke M. Disorders of skeletal muscle, Neurology Clinical Practice, 3rd ed., 2000, 2194-2198.
-
Bushby K. Genetics and the muscular dystrophies. Dev Med Child Neurol. 2000;42(11):780-784.
-
Cifrek M, Medved V, Tonković S, Ostojić S. Surface EMG based muscle fatigue evaluation in biomechanics. Clin Biomech (Bristol, Avon). 2009;24(4):327-340. doi:10.1016/j.clinbiomech.2009.01.010.
-
Dalakas M, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
-
Dubowitz V. Muscle Disorders in Childhood, 2nd ed. Philadelphia, WB Saunders, 1995.
-
Goţia S. Dermatomyositis. In: Moraru E, Ailioaie C (under ed.). Pediatrie. Iaşi, Ed. Vasiliana ‘98, 2008, 159-164.
-
Griggs R, Karparti G. Myoblast Transfer Therapy (Advances in Experimental Medicine and Biology). Dordrecht, Netherlands, Kluwer Academic Publishers, 1990.
-
Guyton A. A Treatise on the Physiology of Man, Bucharest, Callisto, 2007.
-
Jackson CE. A clinical approach to muscle diseases. Seminars in Neurology. 2008;28(2):228-40.
-
Kamen G, Gabriel D. Essentials of Electromyography. Human Kinetics, 2010.
-
Kimura J. Electrodiagnosis in diseases of nerve and muscle: principles and practice, 3rd Ed., New York: Oxford University Press, 2001.
-
Loke J, MacLennan DH. Malignant hyperthermia and central core disease: disorders of Ca2+ release channels. Am J Med. 1998;104(5):470-486.
-
McMillan HJ, Kang PB. Pediatric Electromyography. Springer, 2017.
-
Miller R, Layzer R, Mellenthin M, et al. Emery-Dreifuss muscular dystrophy with autosomal dominant transmission, Neurology. 1985;35(8):1230-1233.
-
Monnier N, Romero N, Lerale J, et al. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum Mol Genet. 2000;9(18):2599-2608.
-
Orban K. Electromyography. Practical Physiology. 2010;61-70.
-
Pitt M. Neurophysiological strategies for the diagnosis of disorders of the neuromuscular junction in children. Dev Med Child Neurol. 2008;50:328-333.
-
Pitt M. Paediatric electromyography in the modern world: a personal view. Dev Med Child Neurol. 2011;53(2):120-124.
-
Popescu V. Neurologie pediatrică. Vol. II. Teora. 2002, 1720-1737.
-
Ramanan AV, Feldman BM. Clinical features and outcomes of juvenile dermatomyositis and other childhood onset myositis syndromes. Rheum Dis Clin North Am. 2002;28(4):833-857.
-
Ranga V, Teodorescu I. Anatomia şi fiziologia omului. Bucharest, Ed. Medicală, 1970.
-
Rider L, Miller F. Idiopathic inflammatory muscle disease: clinical aspects. Baillieres Best Pract Res Clin Rheumatol. 2000;14(1):37-54.
-
Stamatoiu I, Asgian B, Vasilescu C. Electromiografie clinică. Bucharest, Ed. Medicală, 1981.
-
Thrush PT, Allen HD, Viollet L, Mendell JR. Re-examination of the electrocardiogram in boys with Duchenne muscular dystrophy and correlation with its dilated cardiomyopathy. Am J Cardiol. 2009;103(2):262-265. doi:10.1016/j.amjcard.2008.08.064.
-
Turner C, Hilton-Jones D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry. 2010;81(4):358-367. doi:10.1136/jnnp.2008.158261.
-
Wallgren-Pettersson C, Clarke A, Samson F, et al. The myotubular myopathies: differential diagnosis of the X-linked recessive, autosomal dominant, and autosomal recessive forms and present state of DNA studies. J Med Genet. 1995;32(9):673-679.
-
Wattanasirichaigoon D, Swoboda KJ, Takada F, et al. Mutations of the slow muscle alpha-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy. Neurology. 2002;59(4):613-617. doi:10.1212/wnl.59.4.613.
-
https://clinicalgate.com/approach-to-pediatric-electromyography/
-
https://clinicalgate.com/congenital-myopathies/
-
https://www.medicalnewstoday.com/articles/187618.php
-
Moraru E, Ţarcă E, Crişcov I. Distrofia musculară progresivă – update. Pediatru.ro. 2016; https://www.medichub.ro/reviste/pediatru-ro/distrofia-musculara-progresiva-update-id-407-cmsid-64.
-
http://yassermetwally.com/blog/?p=64
-
https://www.mda.org/disease/becker-muscular-dystrophy
-
https://ghr.nlm.nih.gov/condition/emery-dreifuss-muscular-dystrophy#
Articole din ediţiile anterioare
ACTUALITATI/UP-TO-DATE | Ediţia 1 57 / 2020
Colagenozele la copil – aspecte epidemiologice, clinice, diagnostice şi terapeutice
Claudia Sîrbe, Iulia Szabo, Alina Grama, Dr. Mihaela Spârchez, Simona Rednic
Colagenozele sunt un grup heterogen de boli inflamatorii sistemice caracterizate de prezenţa autoanticorpilor formaţi împotriva antigenelor...
19 martie 2020
STUDII CLINICE | Ediţia 4 / 2016
Distrofia musculară progresivă - update
Prof. dr. Evelina Moraru, Elena Ţarcă, Irina Crișcov
Distrofia musculară progresivă Duchenne a fost descrisă de Guillaume Duchenne de Boulogne în 1868. Este forma clinică cea mai comună la copil, inci...
09 noiembrie 2016
CASE REPORT | Ediţia 1 73 / 2024
Management of gingival hyperplasia of genetic cause (Ullrich muscular dystrophy) using diode laser in pediatric patients: a case report
Diana Monica Preda, Denisa-Iulia Dănilă, Roxana Cozubaş, Alexandra Mirică, Alexandra Coroleucă, Cătălin-Ion Chiriac-Babei
În zilele noastre, sistemele laser cunosc o dezvoltare importantă, iar utilizarea lor în stomatologie în general şi în chirurgia orală în mod parti...
03 iunie 2024
PAGINA REZIDENTULUI | Ediţia 1 45 / 2017
Polimiozita - caz clinic
Anca Adam, Prof. dr. Evelina Moraru
Polimiozita este o boală rară din grupul miopatiilor inflamatorii idiopatice, caracterizată prin apariţia la nivelul muşchiului scheletic a unui pr...
04 aprilie 2017