The global incidence of congenital hypoacusis is 1-3 in 1000 live newborns. In the last 20 years, neonatal intensive care units benefited from significant developments in the management of critical newborns, thus raising the number of survivors who are at high risk for neurosensitive hearing loss. Risk factors associated with hearing loss are genetic (Pierre-Robin syndrome, type 2 neurofibromatosis, Down syndrome, DiGeorge syndrome, Beckwith-Wiedemann syndrome, other congenital situations) or non-genetic (cytomegalovirus congenital infection, other in utero infections, admission in the intensive care unit, postnatal infections, head trauma at birth, hypoxic-ischemic encephalopathy, extreme prematurity). All newborns should undergo audiologic testing in the maternity ward to identify hearing loss as soon as possible.
Factori de risc pentru pierderea auzului la nou-născut
Risk factors for hearing loss in the newborn
First published: 28 noiembrie 2022
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/ORL.57.4.2022.7291
Abstract
Rezumat
Incidenţa globală a hipoacuziei congenitale este de 1-3 la 1000 de nou-născuţi vii. În ultimii 20 de ani, secţiile de terapie intensivă neonatală au cunoscut o dezvoltare extraordinară privind managementul nou-născutului critic, crescând numărul supravieţuitorilor care prezintă risc pentru pierderea auzului de tip neurosenzorial. Factorii de risc asociaţi pierderii auzului sunt genetici (sindrom Pierre-Robin, neurofibromatoză de tip 2, sindrom Down, sindrom DiGeorge, sindrom Beckwith-Wiedemann, alte situaţii congenitale) sau nongenetici (infecţie congenitală cu citomegalovirus, alte infecţii in utero, internarea în terapie intensivă neonatală, infecţii postnatale, traumatisme craniene la naştere, encefalopatie hipoxic-ischemică, prematuritate extremă). Toţi nou-născuţii ar trebui să fie testaţi audiologic în maternitate pentru a depista cât mai devreme posibil pierderea auzului.
Introducere
Nou-născuţii internaţi în unităţile de terapie intensivă neonatală au risc crescut de a dezvolta pierderi de auz. Chiar şi diminuarea uşoară sau pierderea unilaterală a auzului pot cauza întârzieri semnificative. Patologia trebuie depistată la timp, pentru a preveni întârzierea de limbaj, comunicare şi dezvoltare cognitivă cauzate de hipoacuzie.
Incidenţa globală a hipoacuziei congenitale este de 1-3 la 1000 de nou-născuţi vii. În ultimii 20 de ani, secţiile de terapie intensivă neonatală au cunoscut o dezvoltare extraordinară privind managementul nou-născutului critic, crescând numărul supravieţuitorilor care prezintă risc (20-40 din 1000 de nou-născuţi) pentru pierderea auzului de tip neurosenzorial.
Factorii de risc asociaţi pierderii auzului sunt:
A. Genetici – aproximativ 50% din pierderile de auz congenitale sunt de origine genetică, transmiterea fiind autozomal-recesivă, dominantă sau X-linkată. Surditatea poate fi izolată sau în cadrul unui sindrom. Se cunosc peste 400 de sindroame genetice care cuprind surditatea, dintre acestea cele mai frecvente fiind:
-
Sindromul Pierre-Robin
-
Neurofibromatoza de tip 2
-
Trisomia 21 sau sindromul Down
-
Sindromul DiGeorge
-
Sindromul Beckwith-Wiedemann.
Alte situaţii congenitale:
-
Istoria familială a pierderii permanente a auzului în copilărie.
-
Şuviţe de păr alb asociate cu sindroame care includ pierderea neurosenzorială permanentă de tip conductor a auzului.
-
Anomalii cranio-faciale, inclusiv cele care interesează pavilionul urechii, canalul auditiv, papiloame auriculare, fosete auriculare sau anomalii ale osului temporal.
-
Neurofibromatoza, boli neurodegenerative.
B. Nongenetici – surditatea poate fi secundară unei leziuni apărute în timpul dezvoltării sistemului auditiv intrapartum sau în perioada perinatală. Aceste leziuni pot să apară în următoarele situaţii:
-
Infecţia congenitală cu citomegalovirus (CMV) – cea mai comună cauză de surditate neurosenzorială nonereditară. Majoritatea (90%) copiilor născuţi cu infecţie cu CMV nu prezintă semne clinice de infecţie, însă pierderea auzului apare la 10-15% dintre aceşti nou-născuţi şi de obicei boala este progresivă. Diagnosticul rapid al infecţiei congenitale este esenţial pentru începerea tratamentului oral cu valganciclovir (înainte de 1 lună de viaţă şi administrat timp de şase luni). Acest tratament este asociat cu ameliorarea funcţiei auditive pe termen lung, având ca rezultat îmbunătăţirea dezvoltării neurologice la 2 ani de viaţă. Este recomandată testarea de screening pentru CMV din urină şi salivă la toţi nou-născuţii care nu au trecut testul audiologic în maternitate.
-
Alte infecţii in utero – herpes, rubeolă, sifilis sau toxoplasmoză. Subliniem importanţa testării tuturor gravidelor pentru infecţii din grupul TORCH (toxoplasmoză, rubeolă, citomegalovirus, herpes simplex, alte infecţii, precum sifilis, hepatită B, virus varicelo-zosterian, parvovirus B19) şi a evitării/prevenţiei contactului cu aceşti patogeni.
-
Internarea în terapie intensivă neonatală pentru mai mult de cinci zile sau pentru oricare dintre următoarele motive, indiferent de durata internării: oxigenare extracorporală cu membrană, ventilaţie asistată, expunere la medicamente ototoxice (gentamicină, tobramicină, amikacină), diuretice de ansă (furosemid) şi hiperbilirubinemie ≥20 mg/dl.
-
Infecţii postnatale (bacteriene sau virale) şi meningite neonatale asociate cu surditate neurosenzorială.
-
Traumatisme craniene la naştere: fracturi ale bazei craniului sau oaselor temporale.
-
Encefalopatie hipoxic-ischemică, la care se adaugă criteriul de hipotermie terapeutică.
-
Prematuritate extremă: imaturitatea tuturor sistemelor şi organelor, internare de lungă durată, risc de osteopenie (rahitism), risc pentru otită medie recurentă sau persistentă în primii doi ani de viaţă.
Prezentare de caz
Prezentăm cazul unui nou-născut de sex masculin, provenit din sarcină cu risc (mama a prezentat infecţii de tract urinar în trimestrele II şi III, pentru care a urmat antibioterapie cu cefuroxim şi amoxicilină-acid clavulanic, exsudat nazal pozitiv pentru Streptococcus aureus meticilino-sensibil, membrane rupte cu 7 ore anterior naşterii), născut spontan, în prezentaţie craniană, la vârsta gestaţională de 38-39 de săptămâni, cu greutate la naştere de 3320 g, scor Apgar 8. Încă de la naştere au fost remarcate multiple anomalii: facies asimetric cu comisură bucală deviată spre dreapta, palatoschizis, microglosie, microretrognaţie, urechi jos implantate, la nivelul urechii drepte trei pediculi preauriculari, la nivelul urechii stângi doi noduli preauriculari, conformaţie particulară cu conduct auditiv extern obstruat, fosetă rudimentară la nivel genian stâng, fren lingual restrictiv scurt, fosetă sacrată. S-a ridicat suspiciunea de sindrom Pierre-Robin, forma sindromică.
În contextul culturilor pozitive recoltate la naştere (cultură tegumente şi coprocultură pozitive pentru Enterococcus durans) şi al probelor inflamatorii pozitive (hemogramă cu leucocitoză cu neutrofilie şi proteină C reactivă crescută) a fost iniţiată antibioterapia cu meropenem şi amikacină intravenos, în doze adaptate perioadei neonatale, cu evoluţie favorabilă şi cu remiterea sindromului biologic inflamator.
Imediat după naştere s-a instituit perfuzie endovenoasă de reechilibrare hidroelectrolitică şi acido-bazică. S-a administrat formulă de lapte prin gavaj orogastric, cu toleranţă digestivă bună, permiţând creşterea graduală a cantităţii de lapte pentru a acoperi nevoia de bază. S-a încercat alimentaţia la biberon, însă nou-născutul a prezentat dificultate în coordonarea respiraţiei-deglutiţiei.
Din punct de vedere respirator, pacientul a menţinut SpO2 în aer atmosferic la 98-100%, cu desaturări în perioadele de agitaţie, prezentând sindrom funcţional respirator uşor-moderat, remarcându-se tirajul suprasternal coroborat cu malformaţia orofaringiană, fără modificări stetacustice pulmonare. A fost echilibrat cardiovascular şi digestiv.
Au fost efectuate multiple consulturi interdisciplinare şi investigaţii paraclinice. Ecografia transfontanelară a fost în limite normale, aparent fără anomalii structurale. Ecografia abdominală a evidenţiat dilataţie pielocaliceală moderată bilaterală. Ecografie cardiacă: canal arterial persistent de talie mică, foramen ovale patent cu şunt stânga-dreapta, variantă anatomică de emergenţă artera coronară stângă. Consultul de neurologie pediatrică remarcă particularităţile constituţionale şi adaugă elemente noi: model de mişcare sacadat, cap în retroaducţie la tracţiune, hipotonie axială, la suspendarea axilară membre inferioare extinse, se susţine ghemuit în decubit ventral, postură neonatală. Consultul oftalmologic identifică hernia ţesutului adipos retroocular extern şi fund de ochi cu aspect normal. Consultul ORL menţionează gnatopalatoschizis, micrognatism, atrezia de conduct auditiv extern stâng şi menţine suspiciunea de sindrom Pierre-Robin.
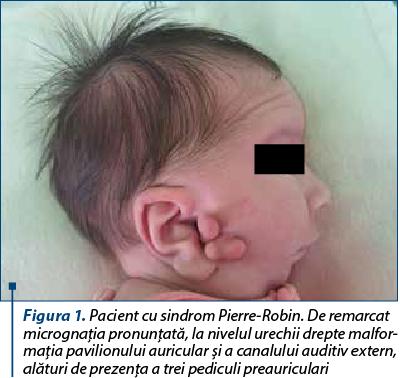
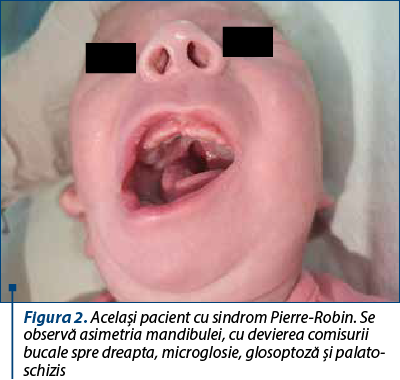
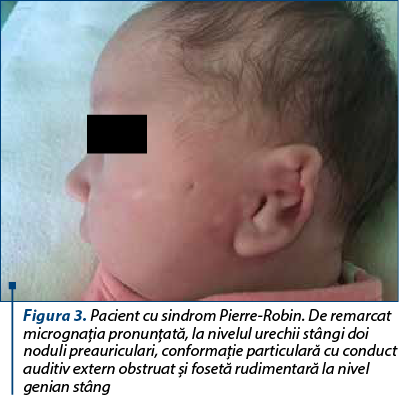
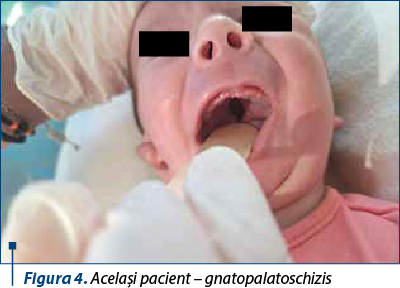
Pentru acest pacient s-a avut în vedere transferul într-o altă unitate sanitară pentru instituirea tratamentului chirurgical (montare traheostomă) şi completarea consulturilor interdisciplinare cu genetică medicală, însă în a 18-a zi de viaţă prezintă alterarea stării generale, sindrom de detresă respiratorie cu tiraj intercostal, subcostal, suprasternal, polipnee (70-80 de respiraţii pe minut), desaturări până la SpO2 50%, bradicardie extremă şi convulsii neonatale. În acest context se decide şi se practică intubaţie orotraheală cu sondă endotraheală şi ventilaţie mecanică. În evoluţie, prezintă detubări repetate din cauza malformaţiilor din sfera ORL şi se montează sondă endotraheală cu balon pentru a evita detubarea accidentală.
În ciuda eforturilor echipei de neonatologie şi a tratamentului instituit (perfuzie de reechilibrare hidroelectrolitică şi acido-bazică, ventilaţie mecanică, antibioterapie, anticonvulsivante de linia a doua şi a treia, transfuzie de masă eritrocitară), pacientul a continuat să prezinte convulsii generalizate confirmate pe înregistrare continuă aEEG (amplitude-integrated EEG), cu degradarea continuă a stării generale până la exitus, în a 23-a zi de viaţă.
Sindromul Pierre-Robin este caracterizat prin secvenţa Pierre-Robin în cadrul căreia sunt prezente micrognaţia şi glosoptoza. Unii pacienţi asociază palatoschizis. Prevalenţa acestui sindrom este estimată la 1 la 8.500-14.000 de persoane. Sunt descrise două forme: nonsindromică şi sindromică, fiecare dintre ele fiind asociată cu mutaţii genetice variate. Alte posibile malformaţii asociate sunt: oftalmice (10-30% dintre pacienţi), cardiovasculare (prevalenţă de 5-58%), musculoscheletale (cele mai frecvente, la 70-80% din cazuri), afectarea sistemului nervos central (întârziere de limbaj sau de dezvoltare, hipotonie, epilepsie, hidrocefalie), genitourinare (testiculi necoborâţi, hidrocel, hidronefroză).
Tratamentul în cazul pacienţilor cu sindrom Pierre-Robin se axează pe menţinerea viabilităţii tractului respirator superior. Tratamentul conservator, cu rezultate bune la pacienţii cu secvenţă Pierre-Robin izolată, presupune urmărirea acestora şi consiliere privind soluţii la dificultăţile de alimentaţie (tehnici de alimentare, alimentarea prin sondă nazogastrică, montarea unei gastrostome). La pacienţii cu micrognaţie pronunţată şi detresă respiratorie severă, poate fi necesar tratamentul chirurgical. De-a lungul timpului au fost descrise şi utilizate multiple tehnici chirurgicale, însă montarea unei traheostome rămâne cea mai utilizată. Managementul pacienţilor cu sindrom Pierre-Robin necesită implicarea unei echipe pluridisciplinare (neonatolog, ORL, ortodont, chirurg maxilofacial, dentist, audiolog, oftalmolog, genetician, pediatru). Mortalitatea variază între 1,7% şi 65%, în funcţie de malformaţiile asociate (malformaţii cardiace, de sistem nervos central).
În concluzie, indiferent dacă au sau nu factori asociaţi, toţi nou-născuţii ar trebui testaţi în maternitate pentru a depista cat mai devreme posibil pierderea auzului. Testarea audiologică a nou-născuţilor este posibilă în maternitate prin includerea în programul naţional de screening. În plus, Joint Committee on Infant Hearing (JCIH) recomandă reevaluarea audiologică la 24-30 de luni de viaţă (recomandare actuală).
Conflicts of interests: The authors declare no conflict of interests.
Bibliografie
-
Hansen AR, Eichenwald EC, Stark AR, Martin CR. Cloherty and Stark’s Manual of Neonatal Care, 8th Edition, Lippincott Williams Wilkins, 2017, 993-996.
-
Smith RJH, Adrian Gooi A. Hearing loss in children: Etiology, 2022, UpToDate (https://www.uptodate.com).
-
Buchanan EP. Syndromes with craniofacial abnormalities, 2022, UpToDate (https://www.uptodate.com).
-
Varadarajan S, Balaji TM, Raj AT, et al. Genetic Mutations Associated with Pierre Robin Syndrome/Sequence: A Systematic Review. Mol Syndromol. 2021;12(2):69-86. doi:10.1159/000513217.
-
Pierre Robin Sequence. Available at: https://rarediseases.org/rare-diseases/pierre-robin-sequence/
-
Tewfik TL, Lacroix Y, Trinh N. Pierre Robin Syndrome. Available at: https://emedicine.medscape.com/article/844143-overview?icd=ssl_login_success_221022#a1
Articole din ediţiile anterioare
The stealthy onset of sudden sensorineural hearing loss in hypothyroidism – literature review
Surditatea neurosenzorială brusc instalată (SSNHL) este o problemă des întâlnită în practica audiologică şi otolaringologică. Pentru a putea ...
Infecţia congenitală cu CMV şi hipoacuzia
Infecţia cu citomegalovirus (CMV) este cea mai obişnuită infecţie congenitală, cu o prevalenţă la naştere între 0,48% şi 1,3% în ultimele decenii...
Evoluţia limbajului post-stroke la vârsta de nou-născut şi sugar
Stroke-ul perinatal este definit ca injurie vasculară cerebrală, care survine până la vârsta de 28 de zile şi include de obicei şi pe cel survenit ...
6 ani de screening auditiv neonatal universal la Iaşi - rezultatele unui parteneriat interdisciplinar
Programul de detecţie a hipoacuziei la nou-născuţi se desfăşoară la Iaşi în mod neîntrerupt din anul 2008. La baza succesului acestui program stă p...