Introduction. Autoimmune hepatitis (AIH) is one of the autoimmune liver disorders recognized in children, along with autoimmune sclerosing cholangitis (ASC) and de novo AIH after liver transplant. This study aimed to assess the treatment in children with AIH, and to identify correlations with disease outcomes. Materials and method. In this retrospective study, we included 30 hospitalized children with AIH, followed-up in our clinic over six years. All patients met the International Autoimmune Hepatitis Group’s simplified diagnostic criteria (IAIHG criteria). Results. Pediatric patients with AIH were aged between 4 months old and 17 years and 8 months old, with a mean age of 6.5 years old. There were 19 girls and 11 boys. Most patients had an acute onset of the disease. Three patients developed acute liver failure. All patients had elevated transaminases and 16 patients had cholestasis. Most patients had increased IgG levels. There were 20 children with type I AIH with SMA and ANA, and 10 children with type II AIH with anti-LKM-1 and anti-LC-1. All patients received prednisone from the diagnosis. Fifteen patients required the addition of azathioprine. Full clinical recovery with a normal level of transaminases was achieved in 18 patients. Complete remission was achieved in 10 patients. Most children had a favorable outcome, with only five patients presenting cirrhosis. Two children died due to complications of autoimmune cirrhosis. Conclusions. The outcome of children with AIH treated with immediate immunosuppressive therapy is favorable, with good long-term survival rates and with a positive effect on the quality of life.
Tratament şi prognostic în hepatita autoimună la copil
Treatment and outcomes of autoimmune hepatitis in children
First published: 31 octombrie 2022
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Pedi.67.3.2022.7236
Abstract
Rezumat
Introducere. Hepatita autoimună (HAI) este una dintre afecţiunile hepatice autoimune recunoscute în populaţia pediatrică, alături de colangita sclerozantă autoimună (CSA) şi de HAI de novo după transplantul hepatic. Scopul acestui studiu a fost de a evalua tratamentul la copiii cu HAI şi de a identifica factorii care influenţează evoluţia bolii. Materiale şi metodă. În acest studiu retrospectiv au fost incluşi 30 de copii spitalizaţi cu HAI, urmăriţi în clinica noastră pe o perioadă de şase ani. Toţi pacienţii au îndeplinit criteriile de diagnostic simplificate ale Grupului Internaţional de Hepatită Autoimună (criteriile IAIHG). Rezultate. Copiii cu HAI au avut vârsta la debutul manifestărilor clinice cuprinsă între 4 luni şi 17 ani şi 8 luni, cu o vârstă medie de 6,5 ani. Au fost 19 fete şi 11 băieţi. Majoritatea pacienţilor au avut un debut acut al bolii. Trei pacienţi au prezentat insuficienţă hepatică acută. Toţi pacienţii au avut transaminaze crescute şi 16 pacienţi au avut colestază. Majoritatea pacienţilor au avut niveluri crescute de IgG. Au fost 20 de pacienţi cu HAI de tip I cu anticorpi antinucleari (ANA) sau anti-muşhi neted (SMA) şi 10 pacienţi cu HAI de tip II cu anticorpi anti-microzom ficat-rinichi (anti-LKM-1) şi/sau anti-citozol hepatic (anti-LC-1). Toţi pacienţii au primit prednison imediat după diagnostic. Cincisprezece pacienţi au necesitat asocierea de azatioprină. Recuperarea clinică completă cu valori normale ale transaminazelor a fost obţinută la 18 pacienţi. Remisiunea completă a fost obţinută la 10 pacienţi. Majoritatea copiilor au avut o evoluţie favorabilă, doar cinci pacienţi prezentând ciroză. Doi copii au murit din cauza complicaţiilor cirozei autoimune. Concluzii. Evoluţia copiilor cu HAI care au fost trataţi cu terapie imunosupresoare imediat după diagnostic este favorabilă, cu rate bune de supravieţuire pe termen lung şi cu un efect pozitiv asupra calităţii vieţii.
1. Introduction
Autoimmune hepatitis (AIH) is one of the autoimmune liver disorders recognized in the pediatric population, along with autoimmune sclerosing cholangitis (ASC) and de novo AIH after liver transplant (LT)(1). AIH is characterized by immune-mediated hepatocytes injury and destruction, causing inflammation, liver failure and fibrosis. AIH is an uncommon disorder described at all ages and in all ethnicities. The increased awareness and the decreased number of cases of viral hepatitis after hepatitis B vaccination and hepatitis C effective treatment have led to more frequently diagnosed AIH compared to the past(1). In contrast, others consider that pediatric AIH incidence has risen in the last two decades(2). AIH can present with wide-ranging clinical symptoms, varying from acute(3) or severe acute hepatitis(4) to chronic hepatitis(5). AIH diagnosis comprises clinical aspects, liver laboratory investigations and immunology analysis, with/without a liver biopsy. Two types of AIH can be distinguished, based on laboratory investigations: type 1 AIH (AIH-1) presents antinuclear antibody (ANA) and/or anti-smooth muscle antibody (SMA), and type 2 AIH (AIH-2) is defined by the presence of anti-liver kidney microsomal type 1 antibody (anti-LKM-1) and/or anti-liver cytosol type 1 antibody (anti-LC-1). AIH diagnosis should be considered after excluding infectious and metabolic disorders(6). Missed diagnosis and late onset of treatment cause progressive inflammatory infiltrate, with continuous loss of hepatocytes, resulting in liver dysfunction and liver fibrosis in the long term(7). The current first-line treatment encompasses prednisolone and azathioprine for clinical and biochemical remission(8). Most patients show complete response to first-line therapy, while some can progress to cirrhosis or liver failure even during remission or relapse after drug withdrawal(9). Hence, further research on the AIH mechanisms and finding novel and effective therapies are essential. Our study aimed to assess the treatment in children with AIH and to identify correlations with disease outcomes.
2. Materials and method
2.1. Patients’ enrolment
This retrospective study included 30 hospitalized children with AIH, followed-up in our clinic over six years (between February 2016 and February 2022), with a mean follow-up period of 3.2 years. All patients fulfilled the simplified diagnostic criteria defined by the International Autoimmune Hepatitis Group (IAIHG criteria)(10). The patients with de novo AIH occurring after liver transplant (LT) and the patients with other liver diseases were excluded. AIH-1 was defined by the presence of SMA and/or ANA, and AIH-2 was defined by LKM-1 or LC-1. Acute liver failure (ALF) was defined as INR>2 and encephalopathy within eight weeks of diagnosis. Typical magnetic resonance cholangiography features defined overlap syndrome of AIH and sclerosing cholangitis (AIH-SC). Cirrhotic AIH was defined based on laboratory investigations and liver stiffness evaluation (fibrosis) on transient elastography (Fibroscan). Remission was defined as the normalization of transaminases and of IgG levels. All patients were started on steroids with the addition of a second agent, depending on the response to steroids. Azathioprine metabolite monitoring was not performed. Children were categorized as AIH-1 versus AIH-2, ALF versus cirrhotic AIH, and overlap sclerosing cholangitis versus non-overlap. The time to follow-up was defined as the date of diagnosis to the most recent follow-up. This study was carried out based on the approval from the Ethics Committee of the Emergency Clinical Hospital for Children Cluj-Napoca, Romania, after receiving informed consent from the parent/guardian.
2.2. Study procedures
The endpoint of this study was the presence of AIH and the adjustment of proper treatment. Demographic data were obtained from patient’s well-documented observation charts, which met the IAIHG criteria. Variables included in the analysis were age, sex, autoantibodies, g-globulins, immunoglobulin A, immunoglobulin G (IgG), immunoglobulin M (IgM) and the absence of viral hepatitis. Autoantibody titers were reported on the basis of local laboratory standards. Liver laboratory tests and specific autoantibodies were carried out upon patient admission to the hospital to obtain the proper diagnosis and to choose the appropriate treatment to achieve remission and improve disease prognosis (survival, transplant or death).
2.3. Measurement of serum markers
The laboratory investigations were performed in the medical lab of the Emergency Clinical Hospital for Children Cluj-Napoca, Romania. Biological samples were collected upon patients’ hospital admission. Patients’ sera were centrifuged right after collection in the lab, then kept at a temperature between -4°C and -20°C.
2.4. Data analysis and statistics
All obtained data were included in a database set up in Microsoft Office Excel and statistically interpreted using the Statistica software, Version 13, TIBCO Software Inc., Palo Alto, CA, USA. The continuous variables were compared by Mann-Whitney U test, and the categorical variables were compared using the Fisher’s exact test. Continuous data were described using median and quartiles. In all analyses, the results were considered significant at p<0.05. For p values below 0.01, we considered the test to have a good statistical significance, while p<0.001 indicated that the statistical significance was extremely important (with an error margin of 0.1%).
3. Results
The pediatric patients with AIH (Table 1) were aged between 4 months old and 17 years and 8 months old, with an average age of 6.5 years old (20 out of the 30 subjects were younger than 12 years old). A female preponderance was observed: 19 girls (63.3%) and 11 boys (36.6%). Eleven patients (36.6%) came from urban areas, and 19 (63.3%) came from rural areas. Only seven patients had family members with autoimmune diseases, such as diabetes, psoriasis and autoimmune thyroiditis, and only one parent had AIH. Associated autoimmune disorders before diagnosis included autoimmune thyroiditis (n=2), IgA deficiency (n=5), immune thrombocytopenic purpura (n=2) and ulcerative colitis (n=1). There were 20 patients with AIH-1 type and 10 patients with AIH-2. There was no significant difference in the duration of illness before diagnosis or frequency of associated autoimmune disorders and family history of autoimmunity between the two types of AIH. Most of the patients had an acute onset, with fatigability (63.3%), loss of appetite (46.6%), nausea and vomiting (40%), abdominal pain, hepato- and/or splenomegaly, followed by jaundice, pruritus, dark urine and pale stools, being indistinguishable from that of acute viral hepatitis. Three out of all patients developed ALF with grade II-IV hepatic encephalopathy. The disease duration before diagnosis had a median of 30 days (12-60 days) for AIH-1, and 22 days (14-90 days) for HAI-2. In 16 patients, there was no history of jaundice. Almost half of the patients (40%) presented the incidental finding of elevated transaminases.
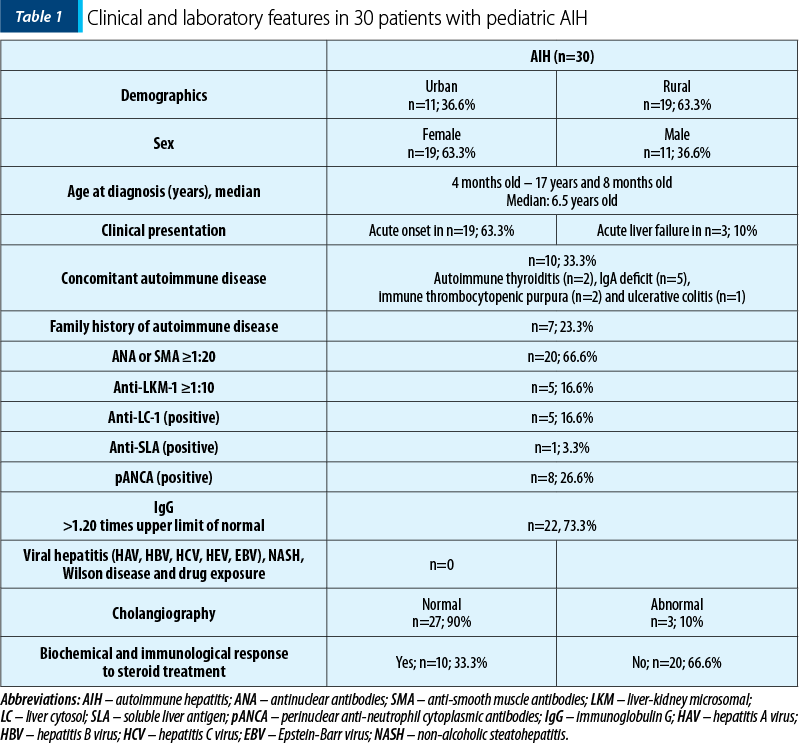
All patients had elevated transaminases and 16 (53.3%) patients presented cholestasis. Most patients (n=22; 73.3%) had elevated IgG levels, but eight children had normal serum IgG level for their age. Partial IgA deficiency was encountered in four patients. Out of 20 children with AIH-1, six were positive for SMA and all were positive for ANA. In the AIH-2 group, half of the patients were positive for LC-1 and half for LKM-1 antibodies. Other antibodies that were present in our cohort were anti-mitochondrial antibodies (AMA; n=2), anti-soluble liver antigen (SLA; n=1) and perinuclear anti-neutrophil cytoplasmic antibodies (p-ANCA; n=8). An overlap syndrome between AIH and ASC was described in three patients.
All patients received prednisolone (or prednisone) at a dosage of 2 mg/kg/day (maximum 60 mg/day) from the moment of diagnosis. During the first weeks of treatment, liver function tests were checked weekly to adjust treatment doses, avoiding severe steroid side effects. In 15 patients, the steroid doses gradually decreased to a maintenance dose of 2.5 to 5 mg/day, after the first 4-8 weeks of treatment, following the improvement of clinical and laboratory analysis. The same 15 patients required the addition of azathioprine as a steroid-sparing agent. Azathioprine was added at a starting dose of 0.5 mg/kg/day in the presence of steroid side effects or if the levels of liver enzymes stopped decreasing on steroid treatment alone. In the absence of signs of toxicity, the dose was increased up to a maximum of 2 mg/kg/day until biochemical control was achieved. Four children had the steroid therapy ceased. Ursodeoxycholic acid (UDCA) treatment was added to immunosuppression in 21 patients.
In our cohort, complete clinical recovery with transaminase levels within the normal range was achieved in 18 patients. IgG levels were normalized in 20 children, and negative or very low-titer autoantibodies were detected in 10 patients. Complete remission was obtained in 10 patients, but only in four patients for more than three years. Eleven patients presented side effects to steroid treatment, with Cushing syndrome, and only two to azathioprine, with pancytopenia. Most children had favorable outcomes, with five patients presenting cirrhosis one to two years after the diagnosis. Two children died, four and, respectively, nine months after the diagnosis, due to complications that appeared in autoimmune cirrhosis.
4. Discussion
This six-year retrospective study of children with AIH provides novel information on the mode of presentation, natural history, treatment and outcomes of this disorder. Because our unit is a tertiary referral center, even though AIH is an uncommon disease, we managed to study and collect all patients’ data from the pediatric AIH cohort of 30 patients.
AIH is described as having female preponderance(11). We described the same preponderance in our AIH cohort. There are few data regarding the incidence of childhood AIH, but it is mentioned that AIH-1 accounts for approximately 60%(12). We showed that two-thirds of children present AIH-1. Most patients presented acute onset of disease indistinguishable from viral hepatitis, although AIH is a chronic liver disease. This corresponds to the findings described in the literature(8). At presentation, AIH could have variable symptoms, and the disease should be suspected in all children with signs and symptoms of prolonged severe liver disease(8). The timing of diagnosis is often delayed in relapsing forms where AIH should be excluded in the presence of sustained symptoms(1). In our study, the disease duration before diagnosis had a median of 3-4 weeks. Incidental finding of raised liver enzymes, without any symptoms or signs, has unknown prevalence(1). In our cohort, almost half of our patients presented the incidental finding of elevated liver enzymes.
The diagnosis of AIH is based on elevated IgG, serum autoantibodies, positive family history for autoimmune disorders, and the exclusion of other causes of pediatric chronic liver disease(1). All patients had elevated liver enzymes, and half of our cases presented cholestasis. Most of the patients had elevated IgG levels. Interestingly, increased levels of IgG were absent in eight children, indicating that normal IgG values do not exclude the diagnosis of AIH in children, which corresponds to the results in the literature(8). Our patients presented serum autoantibodies such as SMA, ANA, LC-1 and LKM-1, AMA, SLA, and pANCA.
The treatment administered to our patients was according to the ESPGHAN Guidelines, which recommend corticosteroid therapy associated or not with azathioprine as the initial medication. The decision of monotherapy or dual therapy belongs to the doctor, depending on the patient’s condition, the center where the patient is treated or on possible contraindications(1) (Figure 1). According to ESPGHAN, up to 75% of children with AIH require both drugs from the start. However, in a recent study, the same group reported a higher incidence of adverse reactions among children who started dual therapy(1).

ESPGHAN recommends corticosteroids for the induction of remission, more precisely prednisolone (or prednisone) which is started with a dose of 2 mg/kg/day (maximum 60 mg/day) and is gradually decreased in 4-8 weeks, according to the level of liver enzymes(13). After that, the maintenance dose is administered for a longer period at the minimum dosage possible, usually 2.5-5 mg/day, depending on the age and weight of the patient. In the first two months, frequent liver laboratory tests are performed to allow proper therapy adjustment and prevent possible drug side effects(14). In these two months, the therapy target is to decrease 80% of liver enzymes, but normal values will be reached only in a few months(8). Although most patients are sensitive to glucocorticoids which represent the cornerstone of AIH therapy, the important challenge appears in the refractory cases or in those who relapse after discontinuing these treatments(15). Our center administered monotherapy with prednisone or prednisolone at starting doses and maintaining doses recommended by the ESPGHAN group.
Azathioprine is the medication usually chosen as a steroid-sparing agent, or it can be started when the value of liver enzymes ceases to decrease on corticosteroids alone. Azathioprine is started at 0.5 mg/kg/day and is increased up to 2-2.5 mg/kg/day, pursuing the normalization of laboratory indexes and preventing any side effects as a treatment goal. Caution is advised in administering azathioprine from the first days of therapy, especially in jaundice, because of its increased liver toxicity(1,16). Usually, the normalization of transaminases levels is obtained after approximately six months(12). The pediatric clinical data regarding initial treatment are still scarce, and further research is needed(17). Fifteen patients in our cohort required the addition of azathioprine as a steroid-sparing agent. According to ESPGHAN recommendations, azathioprine was added at a starting and maintaining dose(1).
Alternative choices of therapy for AIH also include budesonide and calcineurin inhibitors such as cyclosporine and tacrolimus(17). Budesonide presents important hepatic clearance of more than 90% and is clinically better tolerated than prednisolone, but it cannot be used in the presence of cirrhosis(18). Cyclosporine A can be used as induction treatment for remission for six months, followed by standard therapy with prednisone and azathioprine(19). The starting dose of cyclosporine is 4 mg/kg daily, and it can be increased to achieve a blood concentration of 250-300 ng/mL for three months; after that, it can be reduced to 200-250 ng/mL for the next three months, before interrupting it(20). Tacrolimus has similar toxicity as cyclosporine, but it is more efficient for reducing inflammation. There are many debates on using tacrolimus as monotherapy or in combination with prednisolone and/or azathioprine(21). The present experience with budesonide as first-line treatment is limited, and ESPGHAN does not recommend it over the standard treatment(1). We did not use alternative therapies in our pediatric AIH cohort.
Additional second-line treatment options can be used in incomplete remission or in cases with azathioprine side effects: mycophenolate, cyclosporine and tacrolimus(1). Mycophenolate mofetil (MMF) is considered the second most potent immunosuppressive drug after calcineurin inhibitors, but with fewer side effects, making MMF the first drug choice in refractory cases(22). In patients who are intolerant to MMF (diarrhea, nausea, headache and dizziness, and neutropenia), calcineurin inhibitors should be administered(23). New therapeutic methods comprise biological medication and cellular-based therapies. An answer for refractory AIH could stand in the usage of tumor necrosis factor alpha (TNF-a) inhibitors which directly suppress the proinflammatory effects of TNF-a, such as infliximab(24). Another option could be rituximab, a biological therapy based on an anti-CD20 monoclonal antibody that inhibits B lymphocyte proliferation. The successful use of rituximab was reported in only two cases of refractory AIH(25). Recent alternatives promote biological immunotherapy, which could use regulatory T-cell (Treg) proliferation accomplished through genetic engineering(26). Another therapy for refractory AIH cases is sirolimus which intervenes in the regulatory mechanisms of T cells(27). Multicenter prospective research studies based on a larger pediatric population should be performed for more accurate therapy strategies. Clear benefits of anti-CD20 antibodies, anti-TNF-a, and other biological treatments still require further results(1).
Our treatment goals included complete remission, which corresponds to clinical recovery, reduced liver inflammation with normalized transaminases and IgG values, negative or low levels of autoantibodies (ANA and SMA negative or ≤1:20; anti-LKM1 and anti-LC-1 negative or ≤1:10), and the resolution of inflammation on liver biopsy(1). These were the clinical and laboratory criteria for monitoring the treatment response.
Maintenance treatment must be continued for at least two to three years before the withdrawal. After this period, drug cessation should be attempted if the clinical state permits, alongside persistent normal liver tests and IgG levels, and negative autoantibodies (LKM-1) or at low values (ANA/SMA ≤1:10) for 12 months, with no inflammation on histology(1). The cessation of the therapy should not be attempted at puberty because more relapses are reported to occur at this period. Therapy discontinuation is possible in almost one-quarter of patients with AIH-1, while in AIH-2 there are fewer successful attempts(28). The ESPGHAN group recommends gradual withdrawal of prednisolone/prednisone, followed by azathioprine. The chances for successful withdrawal remain low, between 20% and 40%(1). A good therapy response is based on the assessment of IgG levels and on autoantibody titers which can be used to differentiate remission from disease activity(29). Relapse with disease activity is reported in almost 40% of the cases, especially at puberty, due to therapy nonadherence(30). In our cohort, complete clinical recovery with transaminase levels within the normal range was achieved in 18 patients. IgG levels were normalized in two-thirds of the children, with negative or very low-titer autoantibodies in one-third of patients. Complete remission was obtained in ten children in our cohort, but only in four children for more than three years with successful therapy withdrawal.
If the disease is poorly controlled, the inflammatory process could cause liver fibrosis and even cirrhosis(31). In patients with AIH and ALF, liver transplantation (LT) could be a life-saving procedure for patients with decompensated cirrhosis. Recurrence of AIH after LT accounted for 10-50% of patients(32), primarily described in pediatric AIH-2 rather than in AIH-1(33). From 1995 to 2014, liver transplantations were performed for AIH in 3.2% and 3.6% of cases in the United States of America and the United Kingdom, representing an annual rate of 0.84 and, respectively, 0.44 transplants per one million people(34). A five-year survival rate of over 90% is reported in the cases of post-transplant recurrence of AIH(35).
Most children in our cohort had a favorable outcome, with only five patients presenting an unfavorable evolution to cirrhosis in one to two years after the diagnosis and could require LT. Out of these cases, three patients were adolescents who did not administer their medication and did not come to follow-up visits. Also, one patient presented ulcerative colitis E4 in the course of the disease, with an excellent response to the treatment with mesalazine. Unfortunately, the immune liver disease is progressive despite double immunosuppression, and we are currently considering switching to an alternative treatment with MMF. Two children died four and, respectively, nine months after the diagnosis, due to complications that appeared during autoimmune cirrhosis. Eleven patients presented side effects to the steroid treatment, with Cushing syndrome, and only two to azathioprine, with pancytopenia.
5. Conclusions
In this study, we managed to show that the outcome of AIH in children treated with immediate immunosuppressive therapy is favorable, with good long-term survival rates and with a positive effect on the quality of life. End-stage liver disease develops in a few cases, even with accurate therapy, requiring liver transplantation. AIH has a chronic course, implying long-term immunosuppressive treatment, with successful therapy withdrawal in less than a quarter of the cases with AIH-1 and only in a few with AIH-2. The presence of cirrhosis at diagnosis implies a more severe course of the disease, with reduced indexes of short-term and long-term prognosis. Also, an elevated pediatric end-stage liver disease (PELD) score at diagnosis suggests a more resistant disease and usually requires an alternative treatment within the first two years after diagnosis. Despite the research over the last four decades, the complete pathogenesis of AIH is yet to be discovered. The complete understanding of the specific genetic trait, immunoregulatory mechanisms and the interaction between effector and regulatory immunity stay at the ground of the complex and more accurate treatment. Further understanding of disease pathology mechanisms could improve healthcare for patients unresponsive to standard immunosuppression, avoid medication side effects, and prevent possible recurrences after liver transplantation.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Mieli-Vergani G, Vergani D, Baumann U, et al. Diagnosis and Management of Pediatric Autoimmune Liver Disease: ESPGHAN Hepatology Committee Position Statement. J Pediatr Gastroenterol Nutr. 2018;66(2):345-360.
-
Sebode M, Kloppenburg A, Aigner A, et al. Population-based study of autoimmune hepatitis and primary biliary cholangitis in Germany: rising prevalences based on ICD codes, yet deficits in medical treatment. Z Gastroenterol. 2020;58(5):431-438.
-
Czaja AJ. Acute and acute severe (fulminant) autoimmune hepatitis. Dig Dis Sci. 2013;58(4):897-914.
-
Yasui S, Fujiwara K, Yonemitsu Y, et al. Clinicopathological features of severe and fulminant forms of autoimmune hepatitis. J Gastroenterol. 2011;46(3):378-390.
-
Heneghan MA, Yeoman AD, Verma S, et al. Autoimmune hepatitis. Lancet (London, England). 2013;382(9902):1433-1444.
-
Corrigan M, Hirschfield GM, Oo YH, et al. Autoimmune hepatitis: an approach to disease understanding and management. Br Med Bull. 2015;114(1):181-191.
-
Czaja AJ. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J Gastroenterol. 2014;20(10):2515.
-
Gregorio GV, Portmann B, Reid F, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology. 1997;25(3):541-547.
-
Jiménez-Rivera C, Ling SC, Ahmed N, et al. Incidence and Characteristics of Autoimmune Hepatitis. Pediatrics. 2015;136(5):e1237-e1248.
-
Hennes EM, Zeniya M, Czaja AJ, et al. International Autoimmune Hepatitis Group. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology. 2008;48(1):169-76.
-
Tenca A, Farkkila M, Jalanko H, et al. Environmental risk factors of pediatric-onset primary sclerosing cholangitis and autoimmune hepatitis. J Pediatr Gastroenterol Nutr. 2016;62(3):437-442.
-
Gregorio GV, Portmann B, Karani J, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16-year prospective study. Hepatology. 2001;33(3):544-553.
-
Vergani D, Mieli-Vergani G. Pharmacological management of autoimmune hepatitis. Expert Opin Pharmacother. 2011;12:607–13.
-
Beretta-Piccoli BT, Mieli-Vergani G, Vergani D. Autoimmune hepatitis: standard treatment and systematic review of alternative treatments. World J. Gastroenterol. 2017;23:6030–6048.
-
Czaja AJ. Rapidity of treatment response and outcome in type 1 autoimmune hepatitis. J Hepatol. 2009;51(1):161–167.
-
European association for the study of the liver, EASL clinical practice guidelines: autoimmune hepatitis. J Hepatol. 2015;63:971–1004.
-
Woynarowski M, Nemeth A, Baruch Y, et al. Budesonide versus prednisone with azathioprine for the treatment of autoimmune hepatitis in children and adolescents. J Pediatr. 2013;163(5):1347–1353.e1.
-
Mohammad S. Budesonide as first-line therapy for non-cirrhotic autoimmune hepatitis in children: a decision analysis. Scand J Gastroenterol. 2016;51:753–62.
-
Cuarterolo M, Ciocca M, Velasco CC, et al. Follow-up of children with autoimmune hepatitis treated with cyclosporine. J Pediatr Gastroenterol Nutr. 2006;43:635–9.
-
Franulovic OZ, Rajacic N, Lesar T, et al. Cyclosporine induced biochemical remission in childhood autoimmune hepatitis. Coll Antropol. 2012;36:973–9.
-
Marlaka JR, Papadogiannakis N, Fischler B, et al. Tacrolimus without or with the addition of conventional immunosuppressive treatment in juvenile autoimmune hepatitis. Acta Paediatr. 2012;101:993–9.
-
Fallatah HI, Akbar HO. Mycophenolate mofetil as a rescue therapy for autoimmune hepatitis patients who are not responsive to standard therapy. Expert Rev Gastroenterol Hepatol. 2011; 5:517–522.
-
Efe C, Taii HA, Ytting H, Aehling N, et al. Tacrolimus and mycophenolate mofetil as second-line therapies for pediatric patients with autoimmune hepatitis. Dig Dis Sci. 2018;63:1348–1354.
-
Saitis A, Gatselis N, Zachou K, Dalekos GN. Use of TNFα antagonists in refractory AIH: revealing the unforeseen. J Hepatol. 2013;59(1):197-198.
-
Carey EJ, Somaratne K, Rakela J. Successful rituximab therapy in refractory autoimmune hepatitis and Evans syndrome. Rev Med Chile. 2011;139(11):1484.
-
Longhi MS, Liberal R, Holder B, et al. Inhibition of interleukin-17 promotes differentiation of CD25– cells into stable T regulatory cells in patients with autoimmune hepatitis. Gastroenterology. 2012;142(7):1526–1535.
-
Kurowski J, Melin-Aldana H, Bass L, et al. Sirolimus as rescue therapy in pediatric autoimmune hepatitis. J Pediatr Gastroenterol Nutr. 2014;58:e4–6.
-
Deneau M, Jensen MK, Holmen J, et al. Primary sclerosing cholangitis, autoimmune hepatitis, and overlap in Utah children: epidemiology and natural history. Hepatology. 2013;58(4):1392-1400.
-
Gregorio GV, McFarlane B, Bracken P, et al. Organ and non-organ specific autoantibody titres and IgG levels as markers of disease activity: a longitudinal study in childhood autoimmune liver disease. Autoimmunity. 2002;35:515–519.
-
Kerkar N, Annunziato RA, Foley L, et al. Prospective analysis of nonadherence in autoimmune hepatitis: a common problem. J Pediatr Gastroenterol Nutr. 2006;43:629–634.
-
Czaja AJ. Drug choices in autoimmune hepatitis: Part B – nonsteroids. Expert Review of Gastroenterology & Hepatology. 2012;6(5):617–635
-
Mendes F, Couto CA, Levy C. Recurrent and de novo autoimmune liver diseases. Clinics in Liver Disease. 2011;15(4):859–878.
-
Chai PF, Lee WS, Brown RM, et al. Childhood autoimmune liver disease: indications and outcome of liver transplantation. J Pediatr Gastroenterol Nutr. 2010;(50):295–302.
-
Webb G, Rana A, Akhtar MZ, et al. Examining unmet need: international trends in transplantation for autoimmune liver disease. Hepatology. 2016;63:702A.
-
Jossen J, Annunziato R, Kim H-S, et al. Liver transplantation for children with primary sclerosing cholangitis and autoimmune hepatitis: UNOS database analysis. J Pediatr Gastroenterol Nutr. 2017;64:e83–e87.
Articole din ediţiile anterioare
Manifestări extraarticulare în artrita juvenilă idiopatică
Artrita juvenilă idiopatică (AJI) este o boală reumatismală cronică diferită de artrita reumatoidă a adultului. În prezent reumatismul cronic a...
Aspiraţia de corp străin - prezentare de caz
Aspirația de corp străin la copilul mic reprezintă o urgență medicală. Evoluția depinde de natura și de volumul corpului străin, de posibilitat...
Intervenţii nutriţionale precoce în dislipidemii la vârsta pediatrică și riscul pentru sindrom metabolic
Tulburările metabolismului lipidic reprezintă o patologie relativ frecventă la vârsta pediatrică, existând două etiologii principale: terenul ge...
Evaluarea cazurilor de intoxicaţii cu insecticide inhibitorii de colinesterază la copil: studiu retrospectiv pe 7 ani
În acest studiu retrospectiv au fost evaluaţi 87 de copii internaţi în Centrul regional de toxicologie al Spitalului Clinic de Urgenţă pentru Cop...