In recent years, we have seen an increase in thrombotic events in children, which creates difficulties in early diagnosis, especially in rare locations such as portal vein thrombosis. The most common cause is represented by antiphospholipid syndrome (APS), especially secondary to systemic lupus erythematosus (SLE). The investigation of the prothrombophilic pattern is mandatory in these patients, the association being possible. The therapeutic management is complex, in a multidisciplinary team, and will include, in addition to chronic anticoagulation, an effective immunosuppression to control the degree of activity of the lupus disease.
UP-TO-DATE
Capcane de diagnostic în tromboza copilului
Diagnostic pitfalls in child thrombosis
First published: 31 octombrie 2022
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Pedi.67.3.2022.7233
Abstract
Rezumat
În ultimii ani s-a observat o creştere a evenimentelor trombotice la copii, ceea ce creează dificultăţi în diagnosticul precoce, mai ales în localizările rare, cum ar fi tromboza venei porte. Cea mai frecventă cauză este sindromul antifosfolipidic (APS), în special secundar lupusului sistemic (LES). Investigarea tiparului protrombofilic este obligatorie la aceşti pacienţi, asocierea fiind posibilă. Managementul terapeutic este complex, în cadrul echipei multidisciplinare, şi va include, pe lângă anticoagularea cronică, şi o imunosupresie eficientă pentru a controla gradul de activitate al bolii lupice.
In recent years, the number of thrombosis cases in children caused by antiphospholipid syndrome (APS) has increased. This is a constant challenge for the physician, starting from the atypical onset, similar in expression to other pathologies, to the treatment and evolution to either sustained remission or to complications or relapse, a personalized aspect of the individual immunogenetic response. In most cases, the pathology remains underdiagnosed, especially in the absence of specific classification or diagnostic criteria for children, a risk aspect for the initiation of specific therapy, especially since APS is the consequence of generative autoimmune diseases in about 50% of cases. In addition, 21% of children develop systemic lupus erythematosus (SLE) or a lupus-like syndrome on average sex years after the diagnosis of primary APS. The clinical manifestations depend on the area/organ affected by the thrombosis, so that, in situations with rarer localizations such as the liver, adrenal glands, retina etc., there are real diagnostic pitfalls that can negatively affect the evolutionary prognosis of these patients, sometimes even endangering their lives in the case of a catastrophic APS. We present a suggestive clinical case in which the aforementioned aspects apply and which raised numerous management and diagnostic problems.
Clinical case
A.D.G., female, 16 years old
In the family history, her mother has autoimmune thyroiditis.
History of the disease
In 05.2017, she had an appendectomy with normal postoperative evolution.
After one month (6.08.2017), she was hospitalized for abdominal pain accompanied by fever, hepatomegaly and splenomegaly 2 cm below the costal rim.
The biological investigations revealed: anti-cardiolipin antibodies (aCL – IgM) and anti-b2 glycoprotein 1 (B2GP1), increased values of D-dimers, pancytopenia; liver cytolysis x3 increase, mixed hyperbilirubinemia with moderate values, inflammatory syndrome present; negative serologies for viral hepatitis HBV, HCV, EBV-IgM (IgG positive EBV), CMV and toxoplasma; Quantiferon test – negative; total ANA, anti-ADNdc antibodies – absent; anti-LKM1 antibodies – negative; hypertriglyceridemia.
Echocardiography: mitral prolapse, mitral insufficiency grade II, pericardial fluid (1-1.5 cm), tricuspid reflux grade I-II, no valvular vegetations.
Serial abdominal ultrasounds: macronodular hepatomegaly with portal vein dilatation, homogeneous splenomegaly.
The abdominal computed tomography (CT) showed hepatosplenomegaly with hepatic microabscesses, adenopathy in the hepatic hilum, free minimal intraperitoneal fluid.
Stage diagnosis: sepsis; secondary APS; portal hypertension (HTA).
A possible SLE at onset by incidence depending on age, sex and APS association was also discussed, but did not meet the clinical immunologic diagnostic criteria.
The treatment was initiated with the large spectrum antibiotics, analgesics, anticoagulants, transfusions with erythrocyte concentrate and plasma; given the risk of catastrophic APS, pulse therapy with methylprednisolone was initiated. Under this therapy, the evolution was slowly favorable, with clinical-biological remission. Regarding the imaging investigations, an abdominal MRI revealed three infracentimetric nodular liver lesions with the reduction of the liver and the spleen size.
In 9-10.2017, she was hospitalized again, with abdominal pain, diarrhea and bilious vomiting.
An abdominal ultrasound revealed hepatic hilar inflamed lymph nodes, free fluid in Douglas space and left iliac fossa (FIS), hepatomegaly with steatosis, and splenomegaly (130/52 mm).
The biologic investigations revealed: lymphopenia, Hb=10.1 g/dl, platelets = 78,000/mm3, twice increased transaminases, moderate direct hyperbilirubinemia, CRP=188.52 mg/dl, ESR=58 mm/h, D-dimers=1206.5 ng/ml; positive tests for lupus anticoagulant; anticardiolipin and anti-B2GP1 IgM and IgG antibodies present, with an elevated level.
The laparoscopic surgical exploration indicated non-constrictive adhesive bands around cecum corresponding to appendicular scar, with xanthochromatic peritoneal fluid and negative microbiological exam. She received treatment with anticoagulants and continuous systemic cortisone therapy (Medrol®), which she failed to comply with by taking it intermittently.
The following hospitalization (02.2019) was for abdominal pain, without vomiting, diarrhea or fever.
The clinical exam revealed: H=168 cm, W=76 kg, IMC=26.9 kg/m2, obesity, pale skin, erythematous rash on the palms, significant hair loss, stretch marks on abdomen and back, dorsal acne, mildly painful abdomen in the epigastric region, palpable liver and spleen 2 cm from costal margin, TA=115/75 mmHg, SaO2=98%.
The biological investigations revealed: moderate anemia (9.9 g/dl), WBC 10008/mm3 with 75.8% neutrophiles, PLT=195000/mm3, elevated CRP (140 mg/dl), prolonged PT and APTT, APS profile – positive, ADNdc and AAN antibodies present with increased levels; normal renal function, creatinine clearance, the urinary density was normally but positive urine culture for Escherichia coli. The imaging assessment revealed:
Abdominal ultrasound – focal liver steatosis and splenomegaly.
Abdominal CT – hepatosplenomegaly, pseudonodular liver structure, venous return abnormality – Budd-Chiari syndrome in observation.
Abdominal MRI angiography – mild ectasia of ileal veins; caliber and morphology of suprahepatic veins – normal, except for a 10-mm section, immediately before the origin of VC, at the level of all three branches, with irregular signal, data consistent with partial recanalization of a previous thrombosis at the level of suprahepatic veins; inhomogeneous hepatomegaly, splenomegaly.
The cardiology exam, ECG and troponin echocardiography were normal. Rheumatological examination was also normal: capillaroscopy; C3, C4, ANA, dsDNA, haptoglobin, anti-TPO antibodies. The ophthalmological evaluation did not identify any signs of uveitis. The evaluation of hemostasis and thrombosis objectified the Factor II G2021A mutation, suggestive of hereditary thrombophilia. The continuous treatment with the fractionated heparin (Clexane®) was introduced. The gastroenterological evaluation consisted of the following examinations:
Upper digestive endoscopy – early distal esophageal varices, antral vascular ectasias.
Liver biopsy – regenerative changes of liver parenchyma with dilatation of sinusoids, compatible with chronic Budd-Chiari vascular lesions.
Liver elastography – stage 4 fibrosis.
The patient’s final positive diagnosis was:
1. Secondary antiphospholipid syndrome.
2. Chronic Budd-Chiari syndrome.
3. SLE without renal complication.
4. Hereditary thrombophilia – Factor II G2021A mutation.
5. Obesity.
The treatment consisted in the administration of the anticoagulation with Coumadin® (warfarin) and fractionated heparin (Clexane®), until the normalization of INR, then only the Coumadin® treatment under regular INR monitoring. At the same time, immunosuppression in SLE is associated with hydroxychloroquine. After the initiation of this therapy, an improvement of the abdominal pain syndrome was noted in the evolution, but with the maintenance of the persistent inflammatory syndrome (ESR 22-25 mm/h), hypergammaglobulinemia and positivity for the specific tests of APS and borderline SLE immunoserology (DNAdc, anti-Ro, anti-SSA52 antibodies). It was decided to stop the therapy with hydroxychloroquine and start the therapy with azathioprine (Imuran®) in June 2020. Under this treatment, the tests for APS, the serum level of SLE specific antibodies and the inflammatory syndrome were normalizing. During the 2020-2021 pandemic period, she rarely came to the hospital for checkups, but the clinical and the biological balance normalized, without the development of renal complications and with a controlled INR; we mention that the patient didn’t have COVID-19 during the pandemic surveillance (Figures 1, 2 and 3).

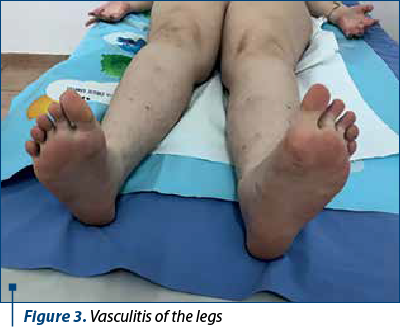
However, from January 2022, the patient interrupted her immunosuppressive treatment and voluntarily increased the dose of anticoagulant without constant INR monitoring, so that in March 2022 she was hospitalized with a severe general condition, fever 40 ºC, skin bruises, ineffective cough, mixed dyspnea, abolition of MV, at the basis of the right posterior hemithorax, abdominal pain predominantly in the right hypochondrium, morbid obesity, vasculitis in the palms and soles, gingival bleeding and intermittent epistaxis. Biologically, there are elements of lupus recurrence associated with severe ASF and INR higher than 10, inflammatory anemia and thrombocytopenia, significant inflammatory syndrome, hypergammaglobulinemia, liver cytolysis x2 increase, high serum level of ferritin, but with normal renal function tests; also, there was associated a radiological appearance of right basal pneumonia with parapneumonic pleuritis. She received broad spectrum antibiotic therapy (Targocid® and gentamicin), antifungals, pulstherapy with methyl-prednisolone followed by systemic cortisone administration (0.25 mg/kg/day) associated with azathioprine, which was resumed, transfusions with erythrocyte mass and liver protection. The dose of anticoagulants was adjusted until the INR was under control. The clinical and biological evolution was slowly favorable over a period of two weeks, after which the patient was monitored by the adult rheumatology clinic, according to her age.
Discussion
The presented case, by the multitude of peculiarities it presents, brings up the complexity of the expression of lupus in children, which leads to difficulties in diagnosis and treatment. The patient, with no personal pathologic history, but with a positive family history for autoimmune disease, developed Budd-Chiari syndrome after appendectomy, initially associated with primary APS and thrombophilia on the F2 mutation, so that later in development it was classified as secondary APS by identifying positivity of specific markers for SLE, but without renal damage. The clinical signature of APS (thrombosis) is associated with an annual incidence rate of 3.1% to 6.6% in children(1,2,3) and mainly affects young children in its primary form, compared with the secondary form, which is more common with age and has a higher incidence than in adults, 60% of cases(4). The clinical manifestation of thrombosis depends on the anatomical area affected – most commonly limbs (40%), lungs and brain (7%), the caliber of the vessels involved – often large to small (6%), and their type. Thus, venous thrombosis is found in more than 60% of cases, while arterial thrombosis accounts for 32%, and mixed thrombosis is found in 2% of cases. Portal vein thrombosis is much rarer (3% of cases) and has the clinical presentation of the Budd-Chiari syndrome, which may have manifestations of acute liver disease, but may also have less suggestive, chronic, recurrent clinical features: more or less localized abdominal pain syndrome such as hepatic pain, dyspeptic syndrome, liver cytolysis of varying degrees, hepatosplenomegaly of varying size. There are a variety of etiologies that cause secondary APS in children, such as autoimmune diseases, neoplasms, infections, medications, but the frequency of SLE (50-83%) is the highest. However, not all patients with SLE experience complications from thrombosis. It appears that the risk of a thrombotic event is significantly higher in lupus patients with a history of vasculitis, avascular necrosis, or positive antiphospholipid antibodies (aPL). A prospective study conducted in 56 children with lupus found that those with positive aPL had a threefold higher risk of organ damage as assessed by the SLICC/ACR Damage Index (SDI)(2,5). Compared with adults, children with lupus and aPL tend to have more active disease at both diagnosis and long-term follow-up. A study comparing 67 SLE children with 131 adults found that the disease activity index (SLEDAI) was significantly higher in children compared with adults, both at diagnosis (16.8 versus 9.3; p<0.0001) and at the average three-year follow-up (5.7 versus 4.6; p=0.012). Even when the disease activity score is normalized, the presence of aPL leads to a higher risk of “disease-related damage” in evolution(6,7). On the other hand, the severity of APS associated with SLE varied from mild to severe catastrophic forms (CAPS), with a lifetime risk in children of 26% versus 40.2% in adults. CAPS was the first manifestation of APS in 87% of children versus 45.2% in adults, with two-thirds of cases manifesting as primary APS. A distinctive feature is the increased association with infection as a trigger in children (60.9% versus 26.8% in adults), making it necessary to diagnose CAPS when a child has infection and multiple organic dysfunctions(3,8). This aspect predominates in the present case, which was triggered by the long-term interruption of immunosuppression and the association of bacterial pneumopathy. The confirmation of the diagnosis of APS is in accordance with the clinical biology criteria of Sydney, 2006(9), which must be associated with the study of etiology, but also with the thrombophilia profile, including the genetic determination of coagulation factors, in order to determine as early as possible the subtype of APS, and develop the most appropriate treatment. Unfortunately, the clinical criteria are not always overlaid by the specificities of the child; for example, we do not always encounter pregnancy-related morbidities as provided by this established grid for adults, especially in children we find vascular thrombosis, which must occur with a frequency of one episode. The immunoserological markers for APS must have a high titer in at least two determinations and persist for at least 12 weeks. The most specific are anti-cardiolipin antibodies, which have high levels at onset and during the course of the disease, and anti-b2GP1 antibodies, which may sometimes be absent at the onset(3). The rare association with genetic alterations for thrombophilia, as shown for Factor II (prothrombin), increases the risk of thrombosis in addition to that induced by APS. Mutation of the Factor II is the second cause of familial thrombophilia. Its incidence in the Caucasian population is 0.7-4% and 18% in those affected by thrombosis. Consequently, it is not enough to intervene therapeutically in these patients only by chronic anticoagulation but, according to the latest guidelines, the immunosuppressive treatment will also be associated with it(10). The last congress dedicated to antiphospholipid syndrome (the 16th edition, in 2019) established the following lines of therapy for this pathology:
-
Vitamin K antagonists (warfarin), oral anticoagulants, antiplatelet agents.
-
Adjuvant therapies – hydroxychloroquine, which also has an antithrombotic effect, statins, due to their pleiotropic but also anti-inflammatory and antithrombotic effects on endothelial cells and monocytes, and vitamin D (low vitamin D levels correlating with venous and arterial thrombotic manifestations in patients with ASF).
-
Biological agents – rituximab (anti-CD20), belimumab (Ab anti-LB activation factor) and anti-TNF alpha agents.
-
Complement activation inhibitors – anti C5 (eculizumab).
Of course, these therapeutic recommendations must be individualized, taking into account the activity level of the lupus disease. In the case of the patient presented, the optimal control for sustained remission and a decrease in disease activity were achieved by replacing hydroxychloroquine with azathioprine in conjunction with anticoagulants.
In conclusion, antiphospholipid syndrome remains an ongoing challenge that presents diagnostic and therapeutic pitfalls and requires the revision of diagnostic criteria and the child’s own exploration, as well as the adaptation of treatment protocols and guidelines.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Ahluwalia J, Singh S, Naseem S, et al. Antiphospholipid antibodies in children with systemic lupus erythematosus: a long-term clinical and laboratory follow-up status study from northwest India. Rheumatol Int. 2014;34(5):669-673.
-
Descloux E, Durieu I, Cochat P, et al. Paediatric systemic lupus erythematosus: prognostic impact of antiphospholipid antibodies. Rheumatology (Oxford). 2008;47(2):183-187.
-
Madison JA, Zuo Y, Knight JS. Pediatric antiphospholipid syndrome. Eur J Rheumatol. 2019 Dec 3;7(Suppl 1):1-10.
-
Avcin T, Cimaz R, Silverman ED, et al. Pediatric antiphospholipid syndrome: clinical and immunologic features of 121 patients in an international registry. Pediatrics. 2008;122(5):e1100-e1107.
-
Driest KD, Sturm MS, O’Brien SH, et al. Factors associated with thrombosis in pediatric patients with systemic lupus erythematosus. Lupus. 2016;25(7):749-753.
-
Mina R, Brunner HI. Pediatric lupus - are there differences in presentation, genetics, response to therapy, and damage accrual compared with adult lupus?. Rheum Dis Clin North Am. 2010;36(1):53-viii.
-
Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562.
-
Berman H, Rodríguez-Pintó I, Cervera R, et al. Pediatric catastrophic antiphospholipid syndrome: descriptive analysis of 45 patients from the “CAPS Registry”. Autoimmun Rev. 2014;13(2):157-162.
-
Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295-306.
-
Cohen H, Cuadrado MJ, Erkan D, et al. 16th International Congress on Antiphospholipid Antibodies Task Force Report on Antiphospholipid Syndrome Treatment Trends. Lupus. 2020;29(12):1571-1593.
Articole din ediţiile anterioare
REVIEW | Ediţia 2 70 / 2023
Modern treatment methods to reduce mortality and morbidity associated with burns in the pediatric patient
Bogdan Caba, Bianca-Andreea Frunză, Ioana-Cezara Caba, Sidonia Susanu
Arsurile pot avea diverse etiologii şi diferite suprafeţe şi grade. În funcţie de gravitate, se pot însoţi de morbiditate şi mortalitate sem...
30 iunie 2023
GHID | Ediţia 2 46 / 2017
Manifestări extraarticulare în artrita juvenilă idiopatică
Bogdan A. Stana, Prof. dr. Evelina Moraru, Alina Murgu, Constantin Ailioaie, Alice Azoicăi
Artrita juvenilă idiopatică (AJI) este o boală reumatismală cronică diferită de artrita reumatoidă a adultului. În prezent reumatismul cronic a...
12 iunie 2017
PAGINA REZIDENTULLUI | Ediţia 2 46 / 2017
Bronșiectaziile non-fibrochistice
Prof. dr. Evelina Moraru, Bogdan A. Stana, Anamaria Răgoșcă
Bronșiectazia este definită clinic prin dilatația permanentă și ireversibilă a calibrului bronhiilor, asociată cu inflamația și infecția croni...
07 iunie 2017
REVIEW | Ediţia 4 64 / 2021
Actualităţi în diagnosticul hepatitei autoimune la copil
Claudia Sîrbe, Alina Grama, Tudor Lucian Pop
Tipurile de boli hepatice autoimune recunoscute la populaţia pediatrică sunt: hepatita autoimună (AIH), colangita sclerozantă autoimună (...
03 decembrie 2021