Frontotemporal dementia (FTD) classically affects adults in their fifties or sixties, but cases have been reported as early as 30 years old. FTD is a neurodegenerative disorder that comprises changes in behaviour and language deficits with gradual onset and progression.
Aim. The aim of this article is to present the unusual case of a frontotemporal dementia in a 37-year-old male who presented first with difficulties on short-term memory, apathy, prosopagnosia, erratic behaviour, visual and auditory hallucinations and disorientation, symptoms with gradual onset and progression for over a year.
Methodology. Upon neurological examination, there were noted: incomplete spatial agnosia, mild sensitive aphasia, anomia, dyscalculia, involuntary movements (myoclonia, choreoathetotic movements), cognitive impairment, grasping reflexes. The biological examination, the psychological evaluation, lumbar puncture, EEG and MRI were performed. The treatment with memantine and trazodone was initiated.
Conclusions. The diagnosis of FTD is not always easy, many differential diagnostics needing to be excluded. The patient must be reevaluated periodically from the neurologic and psychiatric point of view.
Dificultăţi în diagnosticarea demenţei adultului tânăr – prezentare de caz
Challenges in the differential diagnosis of dementia in young adults
First published: 20 noiembrie 2021
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Psih.67.4.2021.5733
Abstract
Rezumat
Demenţa frontotemporală (DFT) afectează în general adulţii cu vârste de 50-60 de ani, dar au fost raportate şi cazuri în jurul vârstei de 30 de ani. DFT este o afecţiune neurodegenerativă ce include modificări ale comportamentului şi deficite de limbaj cu debut şi evoluţie progresive.
Scop. Obiectivul acestui articol este de a prezenta un caz de demenţă frontotemporală la un bărbat de 37 de ani, care a acuzat iniţial dificultăţi ale memoriei imediate, apatie, prosopoagnozie, comportament dezorganizat, halucinaţii vizuale şi auditive şi dezorientare, simptome cu debut progresiv pe perioada unui an.
Metodologie. La evaluarea neurologică s-au obiectivat agnozie spaţială incompletă, afazie senzitivă uşoară, anomie, discalculie, mişcări involuntare (mioclonii, mişcări coreoatetozice), deficit cognitiv şi reflex de apucare. Au fost efectuate investigaţii precum evaluare biologică, examinare psihologică, puncţie lombară, EEG şi RMN. Mematina şi trazodona au fost iniţiate ca terapie farmacologică.
Concluzii. Diagnosticul de DFT nu este uşor, multe diagnostice diferenţiale necesitând a fi excluse. Pacientul va trebui reevaluat periodic din punct de vedere psihiatric şi neurologic.
Introduction
Frontotemporal dementia (FTD) is a neurodegenerative disorder that primarily affects the frontal and temporal lobes and leads to distinct clinical syndromes. It is one of the leading causes of early-onset dementia, usually seen in the fourth and eighth decades, but cases that begin in the third and ninth decades have been reported. About 60% of cases present in people between 45 and 64 years old, and 10% present in patients under 45 years old. Up to 50% of cases are familial, and 10-15% appear to be associated with an autosomal dominant pattern of inheritance. Mutations in genes encoding proteins such as chromosome 9 open reading frame 72 (C9ORF72) and granulin (GRN) are accountable for the majority of familial FTD cases. The hexanucleotide expansions of C9ORF72 correlates with sporadic cases of amyotrophic lateral sclerosis and frontotemporal dementia. Other mutations associated with familial FTD involve: microtubule-associated protein tau (MAPT), transactive response DNA-binding protein 43 (TDP-43), fused in sarcoma (FUS), valosin-containing protein (VCP), and charged multivesicular body protein 2b (CHAMP2B). The prevalence varies widely, ranging from 4-31 per 100,000 in Europe and 15-22 per 100,000 in the United States of America to 2-9.5 per 100,000 in Japan. According to the FTD consensus criteria, it is classified into three clinical variants: behaviour variant frontotemporal dementia (bvFTD), non-fluent variant primary progressive aphasia (nfvPPA), and semantic variant primary progressive aphasia (svPPA)(1). Often, the last two are included in the same subgroup of language-predominant FTD. The spectrum of presentation also includes other motor syndromes, such as cortico-basal degeneration (CBD), progressive supranuclear palsy (PSP) and motor neuron disease like amyotrophic lateral sclerosis (ALS)(2). The bvFTD variant is characterized by loss of empathy, apathy and disinhibition, which typically precede the cognitive impairment, while the nfvPPA variant is characterized by effortful speech production, agrammatism (omission or misuse of grammar) and apraxia of speech, causing inconsistent speech errors such as insertions, deletions, substitutions and transpositions of speech sounds. The svPPA variant usually presents with anomia (word-finding deficits) due to impairments in semantic knowledge. Thus, the speech, although grammatically correct, becomes slow due to word-finding problems(1). Regarding laboratory examinations, currently there are no established biomarkers for the diagnosis of FTD(7). Structural and functional imaging identifies a distinct pattern of changes that correlate with the predominant symptoms and the clinical findings. In the bvFTD, there is an early degeneration of paralimbic structures of the ventromedial prefrontal cortex, anterior cingulate cortex and anterior insula. In the nfvPPA variant, there is a degeneration of the dominant frontal operculum through the premotor area and the insular cortex, while in the semantic variant, there is atrophy in the anterior temporal pole, which eventually spreads to the frontal areas, insula and anterior hippocampus. Functional imaging using SPECT or PET demonstrates selective frontal and/or anterior temporal reduction in blood flow or slow metabolism(2,4).
Regarding diagnosis, a new classification for cognitive disorders was established in the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5), where FTD is referred to as a frontotemporal neurocognitive disorder (NCD), divided into mild or major NCD, according to its severity, also including a behavioural variant or a language variant, according to the clinical manifestation. A diagnostic of probable frontotemporal NCD is made if there is supporting evidence of genetic mutations (family history or laboratory findings) or neuroimaging, otherwise it is categorised as possible(1). On average, the survival is eight years from the symptoms’ onset, with significantly shorter survival for those with motor neuron disease. To date, there is no treatment for FTD that can prevent the progression of the underlying pathologies. A recent randomized double-blind placebo-controlled study showed no benefit of memantine in the treatment of FTD and there are insufficient data to recommend cholinesterase inhibitors. Selective serotonin reuptake inhibitors (SSRIs) are the first-line pharmacotherapy treatments for FTD, also the use of trazodone is cited as having symptomatic efficiency. Second-generation antipsychotic medications should be used with caution, as patients with FTD are more sensitive to extrapyramidal side effects(2,3).
Case report
In February 2021, a 37-year-old male with recent history of psychiatric illness was brought to the Psychiatric Clinic I of the Cluj County Emergency Clinical Hospital, Cluj-Napoca, Romania, by family members, for disorganized behaviour, visual complex hallucinations, possible complex auditory hallucinations, temporal and spatial disorientation and mixt insomnias. The family members recount the symptoms to have an insidious one-year onset, with a progressive anterograde hypomnesia, social withdrawal, decrease in professional efficiency and false non-recognition of work colleagues. The patient, who had the military rank of major sergeant and worked as a licensed gun security officer, was previously admitted in the same month, for two days, at the Psychiatric Department of the Military Hospital Cluj-Napoca, with the diagnostic of acute psychotic disorder without symptoms of schizophrenia, according to the International Classification of Diseases, Tenth Revision (ICD-10). During the previous admission, he underwent psychological examination and received antipsychotic treatment with risperidone 4 mg/day, lorazepam and tiapride, after which he became psychomotor agitated with a disorganized behaviour and therefore in need of constant observation on an emergency psychiatric unit. According to the family, there was no alcohol or substance abuse. On presentation, the patient’s mental status had the following characteristics.
Appearance
-
Personal identification: alternating cooperative with uncooperative episodes, evasive look, and difficulty in sustaining attention.
-
Behaviour and psychomotor activity: episodes of passive motor negativism without waxy flexibility, mimic and gesture stereotypes.
-
General description: partially neat clothing, perplexed, avoiding eye contact.
Speech
Verbal negativism alternating with episodes of dysarthric, slow, monotonous, mumbled speech, lacking productivity, and soliloquy.
Mood and affect
-
Mood: the patient was unable to describe how he feels.
-
Affect: blunted, flat, with narrow range of expression, difficulty in initiating, sustaining or terminating an emotional response, lack of empathy.
Thinking and perception
Form of thinking
-
Productivity: slow thinking, the patient speaks only when questions are asked.
-
Continuity of thought: tangential, loose associations.
Language impairments: partial coherent speech.
Content of thinking and thought disturbances: difficult to evaluate due to lack of productivity, and verbal negativism.
Perceptual disturbances
Hallucinations and illusions: possible complex auditory hallucinations (episodes of soliloquy), possible complex visual hallucinations (the patient had isolated episodes where he sees “flowers” on the floor and “tries to pick them up”).
Depersonalization and derealisation: difficult to evaluate.
Sensorium
Alertness: fluctuation in the level of awareness from preserved vigilance to stupor.
Orientation
-
Time: the patient identifies the year correctly but doesn’t know the exact month and date.
-
Place: the patient knows that he is in a hospital.
-
Person: The patient doesn’t know who the examiner is or the names of the medical staff.
Concentration and calculation: unable to concentrate or resolve mental calculation.
Memory
-
Remote memory: the patient recognises important events occurred when he was younger and free of illness.
-
Recent past memory: impaired memory of the past few months.
-
Recent memory: impaired memory of the past few days. Unable to recall what he did the day before.
-
Immediate retention and recall: unable to repeat all figures after the examiner dictates them, unable to repeat at all backwards.
-
Effect of defect on patient: the patient hasn’t developed cope mechanisms with the defect.
Fund of knowledge: the patient isn’t capable of functioning at the level of his basic endowment and formal education.
Abstract thinking: absent abstraction capability.
Insight: incapable of recognising the illness.
Judgment: unable to evaluate.
The physical examination did not reveal any pathological findings. From a psychiatric point of view, the aforementioned clinical presentation could appoint either an acute psychotic disorder or catatonia. Concluding from the clinical presentation and the heteroanamnesis from the patient’s family, the disease onset was insidious, over a one-year period. The complex visual and auditory hallucinations could be a part of the clinical presentation in an acute psychotic disorder. However, due to the onset type, the duration and progression of the symptoms, along with no associated stress factor noted, the progressive cognitive decline, according to the ICD-10 diagnostic criteria, there were not enough points to conclude this diagnosis. Episodes of passive motor negativism, lack of communication and slow repetitive movements could indicate catatonia. Although these signs were present, there was no rigidity, no waxy flexibility and the repetitive movements were described by the neurologist as being choreoathetosic movement. There were not enough criteria met to conclude a diagnosis of catatonia.
The neurological examination revealed temporospatial disorientation, confusion, cognitive dysfunction with memory impairment (biographical and semantic), dyscalculia, incomplete spatial agnosia, elements of receptive aphasia, anomia, dysarthria, hypophonia, bradipsychia and bradylalia. Myoclonus and choreoathetotic movements of the upper limb were observed. Barre test was positive, with slight pronation of the left upper limb. Independent walk was possible, with diminished balance of the left upper limb. Dysmetria was noted in all four limbs, the osteotendinous reflexes were present, asymmetric but without pathological reflexes and normal muscular tonus. The neurological evaluation concluded an encephalopathic syndrome, with sings of frontal and parietal lobe implication/incomplete Gerstmann syndrome.
The psychological evaluation revealed Mini Mental State Examination score of 11 points, Montreal Cognitive Assessment score of 6 points, and Frontal Assessment Battery score of 3 points.
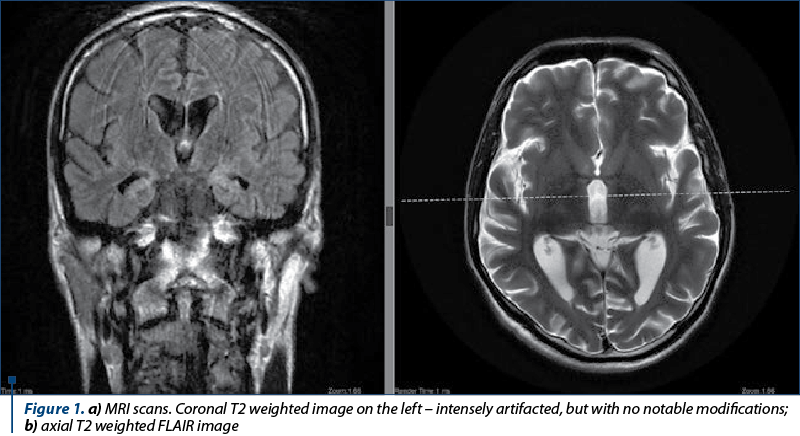
The differential diagnosis involved both psychiatric and neurological disorders, such as: catatonia, acute psychotic disorder, neuroacanthosis, frontotemporal dementia, Wilson’s disease with psychiatric onset, Huntington’s disease and Creutzfeldt Jakob’s disease. Acute psychotic disorder and catatonia were excluded based on the lack of criteria needed for diagnosis according to ICD-10. The routine laboratory findings revealed white blood count 10,260/mm3, C-reactive protein 1.19 mg/dl, folic acid deficit (4.14 ng/dl) and mild deficit of B12 vitamin (174 ng/dl). Serum copper level and ceruloplasmin were within normal range, which ruled out Wilson’s disease. Peripheral blood smear revealed extremely rare acanthocytes and did not indicate neuroacanthosis. HIV and syphilis screening was negative. Serum immunology panel did not reveal any modifications (p-ANCA, c-ANCA, Borrelia IgG + IgM, anti-RO, dc DNA, RNP/SM and antimitochondrial antibodies were all negative) and euthyroidia was noted. An abdominal ultrasound was performed and the imaging of liver, spleen and pancreas had a normal appearance. No focal liver lesions. Gallbladder was normal, with no gallstones demonstrated. No intra- or extra-hepatic duct dilatation. Common bile duct measured 4 mm. Both kidneys had a normal appearance. Native cerebral tomography did not detect any acute haemorrhagic or ischemic lesions. There were three attempts to undergo a magnetic resonance imaging with contrast (Figure 1a and b), due to the marked psychomotor agitation of the patient. Neuroimaging was possible under sedation, but with intense artefacts. Other than diffuse atrophy, no notable lesions or modifications postcontrast administration were observed(6,7).
Electroencephalography (Figures 2, 3 and 4) revealed a background slow rhythm with the predominance of a theta rhythm with 7-8 c/s, in bilateral derivations. On this background, some waves where delta salvos appeared isolated in bilateral frontoparietal derivations. Hypoventilation caused a slowing of the route with the accentuation of the described delta frequency, with the appearance of three-phase waves in the frontal and occipital derivations bilaterally, without a clear periodicity of them (neither on the recorded route, nor only listed). Conclusion: encephalopathic route with the apparent sketching of three-phase, non-periodic waves, without irritative intercritical paroxysmal changes.
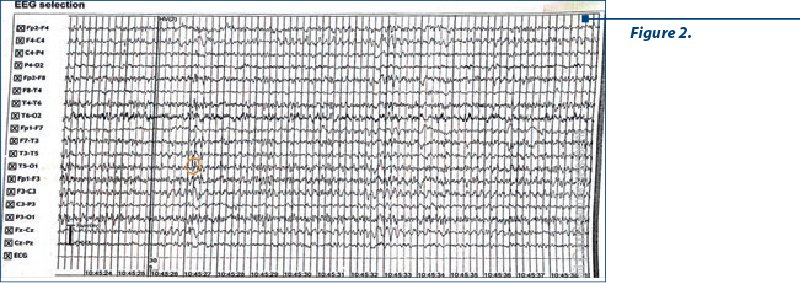
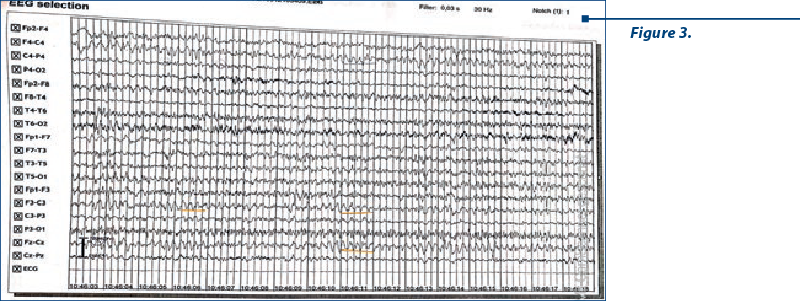
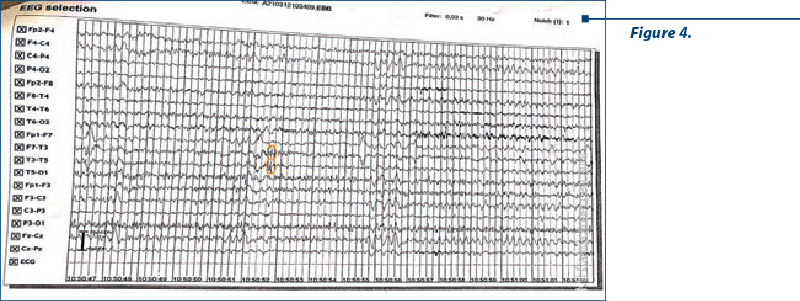
Considering the EEG appearance, a suspicion of Creutzfeld Jakob’s disease was raised. Our patient underwent a lumbar puncture with cerebrospinal fluid analysis: clear, normal pressured fluid was evacuated, and no cells, glucose or cytologic-albumin dissociation were detected. Multiplex panel from the cerebral spinal fluid, including Escherischia coli, Haemophilus influenzae, Neisseria meningitis, Streptococcus pneumoniae, cytomegalovirus, enterovirus, herpes simplex virus 1 and 2, human herpes virus 6 and varicella zoster virus, did not detect the presence of any bacteria or viruses in the cerebrospinal fluid. The 14.3.3 protein, which is patognomic for prion disease, was analysed from the cerebrospinal fluid and the result was negative, which excluded Creutzfeldt‑Jakob’s disease. Considering other hypothesis for early-onset dementia, our patient was also genetically tested for Huntington’s disease, but there was no mutation detected in the huntingtin gene. Excluding the already aforementioned disorders, the psychiatric and neurological signs, negative neuroimaging results, a clinical diagnosis of frontotemporal dementia was made. Considering the family history of the patient (father suffered from amyotrophic lateral sclerosis), a possible genetic correlation with the hexanucleotide expansion of C9ORF72 was made between frontotemporal dementia and amyotrophic lateral sclerosis in our case. However, C9ORF72 gene hexanucleotide repeat expansion testing was not conducted. Based on the diagnostic criteria for frontotemporal dementia, this case fulfils the diagnostic criteria for probable frontotemporal dementia(5,10). The treatment with trazodone and memantine was initiated and the patient remained under psychiatric and neurologic monitoring.
Discussion
This case raised challenges due to the psychomotor agitation the patient presented during the hospitalisation and the imaging investigations. Considering the disease onset was a year ago, the dynamic neuroimaging was necessary and could reveal the classical atrophy of the frontal and temporal lobes. Due to the aforementioned reasons, no enough sequences of imaging could be performed, ergo no volumetric measurements could be assessed on the MRI imaging. The patient remained in observation and SPECT CT will be recommended(9,10). His current neurologic or psychiatric status has not changed and he remained in the care of the family.
Conclusions
The diagnosis of FTD is not always easy, many differential diagnostics needing to be taken under consideration and excluded.
Bibliografie
Sadock B, Sadock V, Ruiz P, Kaplan H. Kaplan and Sadock’s comprehensive textbook of psychiatry. Lippincott Williams & Wilkins. 2009.
Rascovsky K, Hodges J, Knopman D, Mendez M, Kramer J, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456-2477.
Snowden J, Neary D, Mann D. Frontotemporal dementia. Br J Psychiatry. 2002 Feb;180:140-3.
Rascovsky K, Hodges J, Kipps C, Johnson J, Seeley W, Mendez M, et al. Diagnostic Criteria for the Behavioral Variant of Frontotemporal Dementia (bvFTD): Current Limitations and Future Directions. Alzheimer Dis Assoc Disord. 2007 Oct-Dec;21(4):S14-8.
Rohrer J. Structural brain imaging in frontotemporal dementia. Biochim Biophys Acta. 2012 Mar;1822(3):325-32.
Meeter L, Kaat L, Rohrer J, van Swieten J. Imaging and fluid biomarkers in frontotemporal dementia. Nat Rev Neurol. 2017 Jul;13(7):406-419.
Ducharme S, Dols A, Laforce R, Devenney E, Kumfor F, van den Stock J, et al. Recommendations to distinguish behavioural variant frontotemporal dementia from psychiatric disorders. Brain. 2020 Jun 1;143(6):1632-1650.
Chen Y, Kumfor F, Landin-Romero R, Irish M, Piguet O. The Cerebellum in Frontotemporal Dementia: A Meta-Analysis of Neuroimaging Studies. Neuropsychol Rev. 2019 Dec;29(4):450-464.
Häkkinen S, Chu S, Lee S. Neuroimaging in genetic frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis. 2020 Nov;145:105063.
Articole din ediţiile anterioare
Capăt de linie. Studiu privind sinuciderile și tentativele de suicid din reţeaua metroului bucureştean (2008-2021)
Comportamentul suicidar în spaţiul liniilor de metrou are un impact considerabil în rândul publicului călător, martor al acelei tragedii, dar şi în...
„Părinţii mei chiar sunt canibali.“ Schizofrenie paranoidă şi tulburare de personalitate narcisică
Prezentăm cazul unui pacient în vârstă de 35 de ani, diagnosticat cu schizofrenie la vârsta de 22 de ani, cu o personalitate premorbidă dizarmonică...
Actualităţi în terapia tulburării de alimentaţie compulsivă
Tulburarea de alimentaţie compulsivă reprezintă o patologie frecvent întâlnită atât la adolescenţi, cât şi la populaţia adultă, fie ca principal di...
Eficacitatea terapiilor cognitiv-comportamentale din a treia generaţie – o analiză narativă a literaturii
Terapiile cognitiv-comportamentale (TCC) de primă generaţie, apărute în anii 1940, au fost axate pe aspecte observabile în plan acţional, considerâ...